Первичное пигментное узловое заболевание коры надпочечников - Primary pigmented nodular adrenocortical disease
| Первичное пигментное узловое заболевание коры надпочечников | |
|---|---|
| Другие имена | ППНАД |
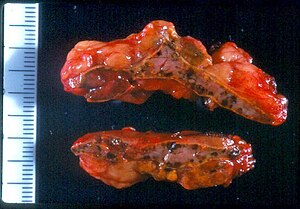 | |
| Макроскопический вид надпочечников в PPNAD после адреналэктомии | |
Первичное пигментное узловое заболевание коры надпочечников (ППНАД) был впервые придуман в 1984 году Карни и др. это часто происходит в связи с Карни комплекс (ЧПУ). CNC - это редкий синдром, который включает образование аномальных опухолей, вызывающих эндокринную гиперактивность.
PPNAD возникает из-за увеличения коры надпочечников, в результате чего синдром Кушинга это не зависит от гормона гипофиза АКТГ.[1]
Признаки и симптомы

PPNAD - редкая причина высокого уровня кортизола в крови и часто проявляется как АКТГ-независимый синдром Кушинга.[2][3] Эффекты PPNAD часто могут быть цикличными, поэтому симптомы синдрома Кушинга не всегда будут такими серьезными, что может затруднить диагностику.[4] Классические симптомы синдрома Кушинга включают быстрое увеличение веса в центре, опухшее красное лицо и буйволиный горб сзади на шее из-за жировых отложений. Кожные изменения при синдроме Кушинга включают легкое истончение и образование синяков, развитие стрии и гиперпигментация кожных складок. Гормональные изменения могут привести к гирсутизм, самцы развиваются ткань молочной железы, у женщин больше нет менструации и оба пола могут стать бесплодными. Высокий уровень кортизола может привести к психологическим расстройствам, таким как беспокойство или депрессия и бессонница. Здоровье костей может ухудшиться, что приведет к повышенный риск перелома у людей с синдромом Кушинга.[5] PPNAD уникален, поскольку он часто вызывает болезнь Кушинга в молодом возрасте, у детей и подростков.[3] В дополнение к другим симптомам синдрома Кушинга, пациент может иметь низкий рост из-за прерывания роста из-за подавления АКТГ.[5]
У 90% людей с PPNAD это связано с Карни Комплекс.[нужна цитата ] Комплекс Карни обычно передается по наследству, но может возникать и спорадически.[6] Видимый признак комплекса Карни - аномальная гиперпигментация кожи. Также может быть миксомы которые могут проявляться в виде комков на коже и груди, а также часто присутствовать в сердце, что может привести к множественным сердечно-сосудистым проблемам.[7] Большинство людей с PPNAD будут иметь некоторые из этих признаков / симптомов из-за тесной связи между PPNAD и комплексом Карни.[нужна цитата ]
Причины
PPNAD, эндокринное проявление, которое происходит от комплекса Карни (CNC), может быть синдромальным или изолированным. Основной причиной изолированного PPNAD является мутация PRKAR1α, расположенная в 17q22-24, который является геном, кодирующим регуляторную субъединицу R1α протеинкиназы A. Гетерозиготные мутации инактивации PRKAR1α зародышевой линии присутствуют у 80% пациентов с ЧПУ, страдающих синдромом Кушинга.[8] Существует более 117 мутаций гена PRKAR1α, которые могут вызывать CNC, причем многие из этих мутаций продуцируют преждевременные стоп-кодоны, что приводит к полной потере белка PRKAR1α. У пациентов с ЧПУ был также обнаружен необычно укороченный белок PRKAR1α, обнаруженный в опухолях и лейкоцитах после мутации сайта сплайсинга, которая вызывает пропуск экзона-6.[9] Следовательно, как гаплонедостаточность, так и полная потеря PRKAR1α могут привести к повышенной активности PKA, наблюдаемой у пациентов с PPNAD, из-за нарушения сигнального пути цАМФ.[нужна цитата ]
Сахут-Барнола и др. использовали мышиную модель для нокаута cre-lox гена Prkar1a, в частности, из клеток коры надпочечников, и наблюдали, что у мышей впоследствии развился синдром Кушинга, который не зависит от гипофиза. Они также заметили, что мутация вызвала повышенную активность PKA.
Потеря R1α привела к тому, что надпочечник взрослого человека стал гиперактивным и гиперпластическим с обеих сторон, поскольку, по-видимому, клетки надпочечника плода внутри него не поддерживались и, таким образом, увеличивались. Это установило опухолевые образования. Эта модель KO мышей фенокопирует то, что происходит в случаях PPNAD у человека.[10]
Инактивация PDE11A4, расположенного в 2q31-5, также была идентифицирована у пациентов с PPNAD без мутаций PRKAR1α. PDE11A4 - это ген, кодирующий фосфодиэстеразу 11A4, еще один участник сигнального пути цАМФ.[11]
Диагностика
Диагноз обычно ставится после исследования причины уже подозреваемого синдрома Кушинга. Высокий уровень кортизола, наблюдаемый у пациентов с PPNAD, не подавляется при введении дексаметазона (тест подавления дексаметазона ), и на МРТ или CT при визуализации гипофиз не покажет никаких отклонений. Измерение АКТГ подтвердит, что причина синдрома Кушинга у пациентов не зависит от АКТГ. Сам по себе синдром Кушинга носит периодический характер, что может затруднить диагностику PPNAD.[12]
Диагноз PPNAD может быть трудно определить до операции, поскольку результаты компьютерной томографии могут быть различными, то есть казаться нормальными или предполагать одностороннее поражение надпочечников, что затрудняет постановку правильного диагноза. НП-59 сцинтиграфия может быть особенно полезно при выявлении двустороннего характера заболевания.[13]
Исследования генов не требуются для диагностики, так как существуют четкие макроскопические и гистологические диагностические маркеры, поскольку узелки обычно отчетливо видны в обоих случаях. Было показано, что положительный семейный анамнез PPNAD связан с аномальными гистологическими данными, например митотические фигуры, которые могут еще больше затруднить диагностику. В точке, где КТ брюшной полости и МРТ гипофизарной ямки не показывают явных аномалий, адреналэктомия может быть выполнено.[12]
лечение
После постановки диагноза важно, чтобы пациенты находились под постоянным наблюдением. Наиболее распространенным методом лечения PPNAD является двусторонняя лапароскопическая адреналэктомия; процесс, при котором оба надпочечника удаляются через небольшой разрез.[2]Пациентам, которые прошли это лечение, будут прописаны минералокортикоидные и глюкокортикоидные стероиды, поскольку они больше не вырабатываются естественным путем.[14]Этот метод лечения используется и совершенствуется с 1984 года.[15]
использованная литература
- ^ Ларсен, Дженнифер Л; Кэти, W.J; Оделл, Уильям Д. (1986). «Первичная узловая дисплазия коры надпочечников, отдельный подтип синдрома Кушинга. Отчет о болезни и обзор литературы». Американский журнал медицины. 80 (5): 976–84. Дои:10.1016/0002-9343(86)90648-0. PMID 3010718.
- ^ а б Чой, Кён Мук; Сеу, Чжэ Хонг; Ким, Ён Хён; Ли, Ын Чжон; Ким, Сан Джин; Байк, Сэй Хён; Чой, Дон Сеоп (1995). «Синдром Кушинга, вызванный первичным пигментным узловым заболеванием надпочечников - клинический случай, обзоры литературы -». Корейский журнал внутренней медицины. 10 (1): 68–72. Дои:10.3904 / kjim.1995.10.1.68. ЧВК 4532033. PMID 7626560.
- ^ а б Куркуцакис, Никос; Прассопулос, Панос; Стратакис, Константин А (2010). «Результаты КТ первичного пигментного узлового заболевания надпочечников: редкая причина АКТГ-независимого синдрома Кушинга». Американский журнал рентгенологии. 194 (6): W541. Дои:10.2214 / AJR.09.4056. PMID 20489078.
- ^ Стратакис, Константин А; Киршнер, Лоуренс С; Карни, Дж. Эйдан (2001). «Клинические и молекулярные особенности комплекса Карни: диагностические критерии и рекомендации по оценке пациента». Журнал клинической эндокринологии и метаболизма. 86 (9): 4041–6. Дои:10.1210 / jcem.86.9.7903. PMID 11549623.
- ^ а б Вагнер-Бартак, Николай А; Байомы, Али; Хабра, Мухаммед Амир; Мухи, Шалини В; Морани, Аджайкумар С; Кориви, Бринда Р.; Вегспак, Стивен Джи; Эльсайс, Халед М (2017). «Синдром Кушинга: диагностическое обследование и особенности визуализации с клинической и патологической корреляцией». Американский журнал рентгенологии. 209 (1): 19–32. Дои:10.2214 / AJR.16.17290. PMID 28639924.
- ^ Стратакис, Константин А; Киршнер, Лоуренс С; Карни, Дж. Эйдан; Пак, Светлана Д; Тайманс, Сьюзан Э; Гиацакис, Христофорос; Чо, Йи Сук; Чо-Чунг, Юн С (2000). «Мутации гена, кодирующего протеинкиназу - регуляторную субъединицу типа I-альфа у пациентов с комплексом Карни». Природа Генетика. 26 (1): 89–92. Дои:10.1038/79238. PMID 10973256. S2CID 36818715.
- ^ Джайн, Соня; Малешевский, Джозеф Дж; Стивенсон, Кристофер Р.; Кларич, Кайл W (2015). «Современная диагностика и лечение миксом сердца». Экспертный обзор сердечно-сосудистой терапии. 13 (4): 369–75. Дои:10.1586/14779072.2015.1024108. PMID 25797902. S2CID 6935363.
- ^ Бертера, Жером (2006). «Комплекс Карни (ЧПУ)». Журнал редких заболеваний Orphanet. 1: 21. Дои:10.1186/1750-1172-1-21. ЧВК 1513551. PMID 16756677.
- ^ Gangoda, L; Doerflinger, M; Шривастава, Р; Нараян, Н. Эджингтон, Л. Э .; Ориан, Дж; Хокинс, C; О'Рейли, Л. А.; Гу, Н; Богио, М; Ekert, P; Штрассер, А; Puthalakath, H (2014). «Потеря Prkar1a приводит к индукции белка семейства Bcl-2 и кахексии у мышей». Гибель клеток и дифференциация. 21 (11): 1815–24. Дои:10.1038 / cdd.2014.98. ЧВК 4211378. PMID 25012505.
- ^ Сахут-Барнола, Изабель; Де Жусино, Сирил; Вал, Пьер; Ламберт-Лангле, Сара; Дэймон, Кристель; Лефрансуа-Мартинес, Анн-Мари; Поинтю, Жан-Кристоф; Марсо, Жоффруа; Сапин, Винсент; Тиссье, Фредерик; Рагаццон, Бруно; Бертера, Жером; Киршнер, Лоуренс С; Стратакис, Константин А; Мартинес, Антуан (2010). «Синдром Кушинга и возрождение особенностей плода у мышей с нокаутом Prkar1a, специфичных для коры надпочечников». PLOS Genetics. 6 (6): e1000980. Дои:10.1371 / journal.pgen.1000980. ЧВК 2883593. PMID 20548949.
- ^ Казабат, Лауре; Рагаццон, Бруно; Груссен, Лайонел; Бертера, Жером (2006). «Мутации PRKAR1A при первичной пигментной узловой болезни коры надпочечников». Гипофиз. 9 (3): 211–9. Дои:10.1007 / s11102-006-0266-1. PMID 17036196. S2CID 95749.
- ^ а б Манипадам, Мариет; Сен, Судипта; Авраам, Рахиль; Саймон, Анна (2011). «Первичная пигментная узловая болезнь коры надпочечников». Журнал Индийской ассоциации детских хирургов. 16 (4): 160–2. Дои:10.4103/0971-9261.86881. ЧВК 3221162. PMID 22121318.
- ^ Веццози, Дельфина; Тененбаум, Флоренция; Казабат, Лауре; Тиссье, Фредерик; Бьенвеню, Мари; Карраско, Кармен А; Лалои-Мишлен, Мари; Барранде, Гаэль; Лефевр, Эрве; Хиеронимус, Сильвия; Табарин, Антуан; Бертанья, Ксавьер; Легманн, Пауль; Вантыгхем, Мари-Кристин; Бертера, Жером (2015). «Гормональная, радиологическая, сцинтиграфия NP-59 и патологические корреляции у пациентов с синдромом Кушинга, вызванным первичным пигментным узловым заболеванием надпочечников (PPNAD)». Журнал клинической эндокринологии и метаболизма. 100 (11): 4332–8. Дои:10.1210 / jc.2015-2174. PMID 26390100.
- ^ Лю, Джеймс К; Флезериу, Мария; Делашоу, Джонни Би; Цирич, Иван С; Кулдвелл, Уильям Т (2007). «Варианты лечения болезни Кушинга после неудачной транссфеноидальной операции». Нейрохирургия. 23 (3): E8. Дои:10.3171 / foc.2007.23.3.10. PMID 17961031.
- ^ Шеной, Б. В; Карпентер, П. С.; Карни, Дж. А (1984). «Двусторонняя первичная пигментная узловая болезнь коры надпочечников. Редкая причина синдрома Кушинга». Американский журнал хирургической патологии. 8 (5): 335–44. Дои:10.1097/00000478-198405000-00002. PMID 6329005.
внешние ссылки
| Классификация | |
|---|---|
| Внешние ресурсы |