Синдром ДиДжорджи - DiGeorge syndrome
| Синдром ДиДжорджи | |
|---|---|
| Другие имена | Аномалия ДиДжорджи,[1][2] велокардиофациальный синдром (VCFS),[3] Синдром Шпринцена,[4] синдром конотрункальной аномалии лица (CTAF),[5] Синдром Такао,[6] Синдром Седлакова,[7] Кардиофациальный синдром Кайлера,[7] СЛОВИТЬ 22,[7] Синдром делеции 22q11.2[7] |
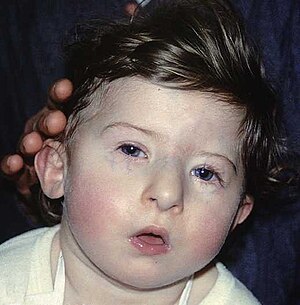 | |
| Ребенок с характерными чертами лица синдрома ДиДжорджи | |
| Специальность | Медицинская генетика |
| Симптомы | Разнообразный; обычно врожденные проблемы с сердцем, особенности лица, волчья пасть[7] |
| Осложнения | Проблемы с почками, потеря слуха, аутоиммунные расстройства[7] |
| Причины | Генетический (обычно новая мутация)[7] |
| Диагностический метод | На основании симптомов и генетическое тестирование[5] |
| Дифференциальная диагностика | Синдром Смита – Лемли – Опица, Синдром Алажиля, ВАКТЕРЛ, Окуло-аурикуло-позвоночный спектр[5] |
| Уход | Включает в себя множество медицинских специальностей[5] |
| Прогноз | Зависит от конкретных симптомов[3] |
| Частота | 1 из 4000[7] |
Синдром ДиДжорджи, также известный как Синдром делеции 22q11.2, это синдром, вызванный удалением небольшого сегмента хромосома 22.[7] Хотя симптомы могут быть разными, они часто включают: врожденные проблемы с сердцем, особенности лица, частые инфекции, отставание в развитии, проблемы с обучением и волчья пасть.[7] Связанные условия включают проблемы с почками, потеря слуха и аутоиммунные расстройства Такие как ревматоидный артрит или же Болезнь Грейвса.[7]
Синдром ДиДжорджи обычно возникает из-за удаления от 30 до 40 гены в центре хромосома 22 в место расположения известный как 22q11.2.[3] Около 90% случаев происходит из-за нового мутация на ранней стадии развития, а 10% - унаследованный от родителей человека.[7] это аутосомно-доминантный, что означает, что для возникновения состояния необходима только одна пораженная хромосома.[7] Диагноз подозревается на основании симптомов и подтверждается генетическое тестирование.[5]
Хотя лекарства и нет, лечение может улучшить симптомы.[3] Это часто включает мультидисциплинарный подход с усилиями по улучшению функции потенциально многих вовлеченных систем органов.[8] Долгосрочные результаты зависят от имеющихся симптомов и серьезности проблем с сердцем и иммунной системой.[3] При лечении продолжительность жизни может быть нормальной.[9]
Синдром ДиДжорджи встречается примерно у 1 из 4000 человек.[7] Синдром был впервые описан в 1968 году американским врачом. Анджело ДиДжорджи.[10][11] В конце 1981 года была определена основная генетика.[11]
Признаки и симптомы
Особенности этого синдрома широко варьируются даже среди членов одной семьи и затрагивают многие части тела. Характерные признаки и симптомы могут включать врожденные дефекты, такие как врожденный порок сердца, дефекты неба, чаще всего связанные с нервно-мышечными проблемами с закрытием (небоглоточная недостаточность ), неспособность к обучению, незначительные различия в чертах лица и повторяющиеся инфекции. Инфекции часто встречаются у детей из-за проблем с иммунная система с Т-клетка -опосредованный ответ что у некоторых пациентов происходит из-за отсутствия или гипопластический вилочковая железа. Синдром ДиДжорджи может быть впервые обнаружен, когда у пораженного новорожденного есть пороки сердца или судороги из-за гипокальциемия из-за неисправности паращитовидные железы и низкий уровень паратироидного гормона (паратгормон ).
У пораженных людей также могут быть другие виды врожденных дефектов, включая аномалии почек и значительные трудности с кормлением в младенчестве. Проблемы с желудочно-кишечным трактом также очень распространены в этой популяции пациентов. Проблемы с перистальтикой пищеварения могут привести к запорам.[12] Такие расстройства, как гипотиреоз и гипопаратиреоз или же тромбоцитопения (низкий уровень тромбоцитов) и психиатрический болезни - частые поздние признаки.[13]
Микроделеции в хромосомной области 22q11.2 связаны с 20-30-кратным повышением риска шизофрения.[14] Исследования показывают различные уровни 22q11.2DS при шизофрении, от 0,5 до 2,0% и в среднем около 1,0%, по сравнению с общим расчетным риском 22q11.2DS в 0,025% в общей популяции.[15]
Существенные особенности можно резюмировать с помощью мнемонической СЛОВИТЬ 22 для описания 22q11.2DS, где 22 означает хромосомную аномалию, обнаруженную на 22-й хромосоме, как показано ниже:[16]
- Сердечная патология (обычно прерванная дуга аорты, артериальный ствол и тетралогия Фалло )
- Аномальный фации
- Тимический аплазия
- Волчья пасть
- Гипокальциемия / гипопаратиреоз
У людей может быть много возможных особенностей, варьирующихся по количеству связанных с ними особенностей от легких до очень серьезных. Признаки распространенности включают:
- Врожденный порок сердца (40% лиц), особенно конотрункальный пороки развития (прерванная дуга аорты (50%), стойкий артериальный ствол (34%), тетралогия Фалло и дефект межжелудочковой перегородки )
- Цианоз (голубоватая кожа из-за плохой циркуляции богатой кислородом крови)
- Небный аномалии (50%), особенно небоглоточная недостаточность, подслизистый волчья пасть, и волчья пасть; характерные черты лица (присутствуют в большинстве Кавказский физических лиц) в том числе гипертелоризм
- Проблемы в изучении (90%), в том числе когнитивные нарушения, расстройства дефицита внимания[17]
- Гипокальциемия (50%) (из-за гипопаратиреоза)
- Существенный кормление проблемы (30%)
- Почечный аномалии (37%)
- Потеря слуха (обе проводящий и нейросенсорная ) (потеря слуха с черепно-лицевыми синдромами )
- Ларинготрахеоэзофагеальные аномалии
- Гормон роста недостаток
- Аутоиммунные расстройства
- Иммунные расстройства из-за сокращения Т-клетка числа
- Судороги (с или без гипокальциемия )
- Скелетный аномалии
- Психиатрические расстройства[17]
Для этого синдрома характерно: неполная пенетрантность. Таким образом, клинические проявления у разных пациентов сильно различаются. Это часто затрудняет раннюю диагностику.[18]
Когнитивные нарушения
Дети с синдромом ДиДжорджи имеют особый профиль в нейропсихологических тестах. Обычно они имеют IQ ниже порогового уровня, при этом большинство людей имеют более высокие баллы в вербальной области, чем в невербальной. Некоторые могут посещать обычные школы, другие учатся на дому или в специальных классах. Тяжесть гипокальциемии в раннем детстве связана с поведенческими трудностями, напоминающими аутизм.[19]
Взрослые с синдромом ДиДжорджи относятся к группе повышенного риска развития шизофрении. Около 30% имеют хотя бы один случай психоз и около четверти развивают фактические шизофрения.[20]
Люди с синдромом ДиДжорджи также имеют более высокий риск развития раннего начала. болезнь Паркинсона (PD). Диагностика болезни Паркинсона может быть отложена на срок до 10 лет из-за использования нейролептики, который может вызвать симптомы паркинсонизма.[21][22]
Речь и язык
Текущие исследования демонстрируют уникальный профиль речевых и языковых нарушений, связанных с 22q11.2DS. Дети часто имеют более низкие оценки речи и языка по сравнению с их невербальными показателями IQ.[противоречивый ] Общие проблемы включают гиперназальность, задержку речи и ошибки звука речи.[23][24][25]
Гиперназальность происходит, когда воздух выходит через нос во время произнесения звуков устной речи, в результате чего понятность. Это общая характеристика речевого и языкового профиля, потому что 69% детей имеют небный аномалии. Если структура мягкого неба велум таков, что он не мешает потоку воздуха подниматься в носовая полость, это вызовет гиперназальная речь. Это явление обозначается как зрительно-глоточная недостаточность (ВПИ). Потеря слуха также может способствовать повышенной гиперназальности, поскольку дети с нарушениями слуха могут испытывать трудности с самоконтролем своей устной речи. Варианты лечения, доступные для VPI, включают протезирование и операцию.[23][24][26][27][28]
Трудности со словарным запасом и формулировкой разговорной речи (выразительный язык дефициты) в начале языкового развития также являются частью речевого и языкового профиля, связанного с делецией 22q11.2. Приобретение словарного запаса у детей дошкольного возраста часто бывает очень поздно. В некоторых недавних исследованиях у детей был очень ограниченный словарный запас или они все еще не вербали в возрасте 2–3 лет. Дети школьного возраста прогрессируют в выразительной речи по мере взросления, но у многих по-прежнему возникают задержки и возникают трудности при выполнении языковых задач, таких как вербальное воспроизведение повествований и создание более длинных и сложных предложений. Восприимчивый язык, то есть способность понимать, удерживать или обрабатывать устную речь, также может быть нарушена, хотя обычно не с такой же серьезностью, как нарушения выразительной речи.[24][27][28][29]
Артикуляция ошибки часто встречаются у детей с синдромом ДиДжорджи. Эти ошибки включают ограниченный фонематический (речевой) инвентарь и использование компенсаторных стратегий артикуляции, что приводит к снижению разборчивости. В фонематический Обычно производимый инвентарь состоит из звуков, издаваемых в передней или задней части ротовой полости, таких как: / p /, / w /, / m /, / n / и глоттальные звуки. Звуки, издаваемые в середине рта, полностью отсутствуют. Компенсирующие артикуляционные ошибки, совершаемые этой группой детей, включают: глоттальные остановки, носовые замены, глоточные фрикативы, лингва-небные свистящие звуки, снижение давления на согласные звуки или сочетание этих симптомов. Из этих ошибок наибольшую частоту встречаемости имеют глоттальные остановки. Обосновано, что ограниченный фонематический инвентарь и использование компенсаторных стратегий артикуляции присутствует из-за структурных аномалий неба. Нарушения речи у этой популяции более серьезны в младшем возрасте и имеют тенденцию к постепенному улучшению по мере взросления ребенка.[23][27]
Генетика

Синдром ДиДжорджи вызван гетерозиготный делеция части длинного плеча (q) хромосомы 22, область 1, группа 1, поддиапазон 2 (22q11.2). Примерно 80-90% пациентов имеют делецию 3 МБ и 8% имеют удаление 1,5 Мб.[30][31] Количество генов, затронутых делецией, составляет примерно от 30 до 50.[32][33] Очень редко у пациентов с подобными клиническими особенностями могут быть делеции на коротком плече хромосомы 10.[34] Заболевание имеет аутосомно-доминантный тип наследования.
Французское исследование 749 человек, которым был поставлен диагноз в период с 1995 по 2013 год, показало, что мутация унаследована у 15% пациентов, из которых 85,5% были от матери.[35] Другие исследования показали, что уровень наследования составляет 6-10%. Большинство случаев являются результатом de novo (новое в семье) удаление.[12] Это связано с тем, что область 22q11 имеет структуру, которая делает ее очень склонной к перестройкам во время образования сперматозоидов или яйцеклеток.[36]
Точный механизм, вызывающий все связанные с этим синдромом признаки, неизвестен.[30] Было установлено, что из 30–50 генов в удаленной области некоторые, возможно, играют роль в развитии некоторых признаков и симптомов.
TBX1
Гаплонедостаточность из TBX1 ген (фактор транскрипции T-box TBX1) считается причиной некоторых наблюдаемых симптомов. Точечные мутации в этом гене также наблюдались у людей с синдромом ДиДжорджи.[30] TBX1 является частью Т-образная коробка семейство генов, которые играют важную роль в формировании тканей и органов во время эмбрионального развития и могут играть роль в регуляции дифференциация пост-миграции клетки нервного гребня. Нервный гребень образует многие структуры, пораженные синдромом ДиДжорджи, включая кости черепа, мезенхима лица и неба, оттока сердца, тимуса и паращитовидной железы строма. Когда происходит потеря выражения FGF18 во время разработки глоточные дуги наблюдается гибель клеток нервного гребня. Хотя ни FGF18, ни TBX1 не экспрессируются в клетках нервного гребня, TBX1 может играть роль в регуляции экспрессии FGF18, обеспечивая правильную дифференцировку этих клеток в глоточной области. Следовательно, дисфункция TBX1 может быть причиной некоторых симптомов синдрома ДиДжорджи.[31]
Исследования на моделях мышей показали, что делеция Tbx1 приводит к нескольким дефектам, аналогичным тем, которые наблюдаются у людей, в основном влияя на развитие великие артерии и вилочковая железа.[37][38]
Аномалии, наблюдаемые в магистральных артериях мышей с дефицитом Tbx1, являются следствием аномального образования и ремоделирования дуги аорты во время раннего развития. Роль Tbx1 в правильном формировании и ремоделировании дуг аорты была широко изучена на различных моделях мышей, что указывает на ключевую роль Tbx1 в развитии сердечно-сосудистой системы и фенотипах, наблюдаемых при синдроме Ди Джорджи.
DGCR8
У мышей гаплонедостаточность DGCR8 ген был связан с неправильной регуляцией микроРНК miR-338 и фенотип с делецией 22q11.2.[39]
TANGO2
Организация транспорта и гольджи 2 гомолог (TANGO2 ), также известный как открытая рамка считывания 25 хромосомы 22 (C22orf25), представляет собой белок, который у человека кодируется геном TANGO2.
Ген, кодирующий C22orf25, расположен на 22-й хромосоме в месте q11.21, поэтому он часто связан с синдромом делеции 22q11.2.[40] Но аутосомно-рецессивное заболевание TANGO2 возникает не во всех случаях.
Мутации в гене TANGO2 могут вызывать дефекты митохондрий. β-окисление[41] и увеличился эндоплазматический ретикулум стресс и снижение Гольджи объемная плотность.[42] Эти мутации приводят к раннему началу гипогликемия, гипераммониемия, рабдомиолиз, сердечные аритмии, и энцефалопатия что позже перерастает в когнитивные нарушения.[41][42]
Гены болезни Паркинсона
22q11.2DS был связан с более высоким риском раннего начала болезнь Паркинсона (PD). Наблюдаемая невропатология похожа на LRRK2 -ассоциированный ПД. Ни один из генов, затронутых у людей с 22q11.2DS, ранее не был связан с БП, но есть несколько вероятных кандидатов. К ним относятся DGCR8, который важен для биогенеза микроДНК мозга, SRPT5 который кодирует белок, который взаимодействует с ПАРК2 белок COMT который участвует в регуляции уровней дофамина, и микроРНК miR-185, которая, как полагают, нацелена на известные локусы PD LRRK2.[21]
Диагностика


Диагностика синдрома ДиДжорджи может быть затруднена из-за количества потенциальных симптомов и различий в фенотипах у разных людей. Это подозревается у пациентов с одним или несколькими признаками делеции. В этих случаях диагноз 22q11.2DS подтверждается наблюдением делеции части длинного плеча (q) хромосомы 22, области 1, полосы 1, поддиапазона 2. Генетический анализ обычно выполняется с использованием флуоресценция на месте гибридизация (FISH), который способен обнаруживать микроделеции, которые стандартное кариотипирование (например, G-полосы ) скучать. Новые методы анализа включают Зависимая от лигирования мультиплексная амплификация зонда анализ (MLPA) и количественная полимеразная цепная реакция (qPCR), оба из которых могут обнаруживать атипичные делеции в 22q11.2, которые не обнаруживаются FISH.[44] Анализ qPCR также быстрее, чем FISH, который может длиться от 3 до 14 дней.[12]
Исследование 2008 года нового зонда MLPA высокого разрешения, разработанного для обнаружения изменение количества копий в 37 точках хромосомы 22q обнаружил, что он так же надежен, как и FISH, в обнаружении нормальных делеций 22q11.2. Он также смог обнаружить небольшие атипичные делеции, которые легко пропустить с помощью FISH. Эти факторы, наряду с более низкой стоимостью и более легким тестированием, означают, что этот зонд MLPA может заменить FISH в клинических испытаниях.[45]
Генетическое тестирование с использованием BACs-on-Beads было успешным в обнаружении делеций, соответствующих 22q11.2DS, во время пренатального тестирования.[46][47] Сравнительная геномная гибридизация (array-CGH) использует большое количество зондов, встроенных в чип, для проверки всего генома на наличие делеций или дупликаций. Его можно использовать в послеродовой и пренатальной диагностике 22q11.2.[48]
Менее 5% людей с симптомами синдрома ДиДжорджи имеют обычные стандартные цитогенетические исследования и отрицательные результаты тестирования FISH. В этих случаях причиной являются атипичные делеции.[49] Некоторые случаи синдрома делеции 22q11.2 имеют дефекты в других хромосомах, особенно делецию в области хромосомы 10p14.[34]
Уход
Лекарства от синдрома ДиДжорджи не существует. Некоторые индивидуальные особенности поддаются лечению с помощью стандартных методов лечения.[50] Ключевым моментом является определение каждой из связанных функций и управление каждой из них с использованием наилучших доступных методов лечения.
Например, у детей важно, чтобы проблемы с иммунитетом выявлялись на ранней стадии, поскольку требуются особые меры предосторожности при переливании крови и иммунизации живыми вакцинами.[51] Трансплантация тимуса может использоваться для устранения отсутствия вилочковой железы при так называемом «полном» синдроме ДиДжорджи.[52] Бактериальный инфекции обращаются с антибиотики. Операция на сердце часто требуется при врожденных пороках сердца. Гипопаратиреоз, вызывающий гипокальциемию, часто требует пожизненного приема витамина D и кальция. Специализированные клиники, которые предоставляют мультисистемную помощь, позволяют пациентам с синдромом ДиДжорджи быть оценены на предмет всех их потребностей в отношении здоровья и позволяют тщательно наблюдать за пациентами. Примером такого типа системы является клиника удаления дефектов 22q по адресу: Больница SickKids в Торонто, Канада, который предоставляет детям с синдромом делеции 22q11 постоянную поддержку, медицинское обслуживание и информацию от группы медицинских работников.[53]
Эпидемиология
По оценкам, синдром ДиДжорджи поражает от одного ребенка из 2000 до одного из 4000 живорождений.[54][55] Эта оценка основана на основных врожденных дефектах и может быть заниженной, поскольку у некоторых людей с делецией мало симптомов и, возможно, им не был поставлен официальный диагноз. Это одна из наиболее частых причин Интеллектуальная недееспособность из-за синдрома генетической делеции.[56]
Ожидается, что число пострадавших вырастет по нескольким причинам: (1) хирургические и медицинские достижения, все большее число людей выживают после пороков сердца, связанных с синдромом. Эти люди, в свою очередь, заводят детей. Шансы на то, что у человека с синдромом ДиДжорджи родится пораженный ребенок, составляют 50% для каждой беременности; (2) Родителям, которые затронули детей, но которые не знали о своих генетических заболеваниях, теперь ставят диагноз по мере появления возможности генетического тестирования; (3) Методы молекулярной генетики, такие как FISH (флуоресцентная гибридизация in situ), имеют ограничения и не позволяют обнаружить все делеции 22q11.2. Новые технологии смогли обнаружить эти нетипичные делеции.[57]
Имя
Признаки и симптомы синдрома Ди Джорджи настолько разнообразны, что разные группы его признаков когда-то считались отдельными состояниями. Эти оригинальные классификации включали велокардиофациальный синдром, синдром Шпринцена, последовательность / синдром ДиДжорджи, синдром Седлаковой и синдром конотрункальной аномалии лица. Теперь все понимают как проявление одного синдрома.
В версии МКБ-10 2015 синдром ДиДжорджи упоминается с использованием двух кодов: D82.1 (синдром Ди Джорджа).[58] и Q93.81 (Велокардио-лицевой синдром).[59] В бета-проекте МКБ-11 синдром обсуждается как «фенотип LD50.P1 CATCH 22».[59] Однако, поскольку этот синдром вызван удалением небольшого фрагмента хромосома 22, некоторые рекомендуют использовать название «синдром делеции 22q11.2 (22q11.2DS)».[60][12] Некоторые эксперты поддерживают изменение названия синдромов ДиДжорджи и велокардиофациального синдрома на CATCH-22.[нужна цитата ] Международный фонд 22q11.2 в рамках своей «Кампании за одноименное имя» защищает синдром делеции имени 22q11.2.[61]
Смотрите также
Рекомендации
- ^ Рапини, Рональд П .; Болонья, Жан Л .; Йориццо, Джозеф Л. (2007). Дерматология: 2-томный набор. Сент-Луис: Мосби. ISBN 978-1-4160-2999-1.
- ^ Джеймс, Уильям Д .; Бергер, Тимоти Дж .; и другие. (2006). Кожные болезни Эндрюса: клиническая дерматология. Saunders Elsevier. ISBN 978-0-7216-2921-6.
- ^ а б c d е «Синдром делеции 22q11.2». Информационный центр по генетическим и редким заболеваниям (GARD). В архиве из оригинала 5 июля 2017 г.. Получено 15 мая 2017.
- ^ Шпринцен Р.Дж., Гольдберг РБ, Левин М.Л., Сидоти Э.Дж., Беркман М.Д., Аргамасо Р.В., Янг Д. (январь 1978 г.). «Новый синдром, включающий волчью пасть, сердечные аномалии, типичные лица и проблемы с обучением: велокардио-лицевой синдром». Расщелина неба J. 15 (1): 56–62. PMID 272242.
- ^ а б c d е «Синдром делеции хромосомы 22q11.2 - NORD (Национальная организация по редким заболеваниям)». NORD (Национальная организация по редким заболеваниям). 2017. В архиве из оригинала 28 января 2017 г.. Получено 10 июля 2017.
- ^ Burn J, Takao A, Wilson D, Cross I, Momma K, Wadey R, Scambler P, Goodship J (октябрь 1993 г.). «Синдром конотрункальной аномалии лица связан с делецией хромосомы 22q11». J. Med. Genet. 30 (10): 822–4. Дои:10.1136 / jmg.30.10.822. ЧВК 1016562. PMID 8230157.
- ^ а б c d е ж грамм час я j k л м п «Синдром делеции 22q11.2». Домашний справочник по генетике. Июль 2013. В архиве из оригинала 13 мая 2017 г.. Получено 15 мая 2017.
- ^ Кобринский Л.Дж., Салливан К.Е. (октябрь 2007 г.). «Велокардиофациальный синдром, синдром Ди Джорджи: синдромы делеции хромосомы 22q11.2». Ланцет. 370 (9596): 1443–52. Дои:10.1016 / S0140-6736 (07) 61601-8. PMID 17950858.
- ^ Гольдман, Ли; Шафер, Эндрю И. (2015). Электронная книга по медицине Goldman-Cecil. Elsevier Health Sciences. п. 702. ISBN 9780323322850. В архиве из оригинала от 05.11.2017.
- ^ ДиДжордж, А (1968). «Врожденное отсутствие тимуса и его иммунологические последствия: сочетание с врожденным гипопаратиреозом». Фонд марша десятицентовиков-врожденных дефектов: 116–21.
- ^ а б Restivo A, Sarkozy A, Digilio MC, Dallapiccola B, Marino B (февраль 2006 г.). «Синдром делеции 22q11: обзор некоторых аспектов биологии развития сердечно-сосудистой системы». J Cardiovasc Med (Хейгерстаун). 7 (2): 77–85. Дои:10.2459 / 01.JCM.0000203848.90267.3e. PMID 16645366.
- ^ а б c d Макдональд-МакГинн Д.М., Салливан К.Э. (январь 2011 г.). «Синдром делеции хромосомы 22q11.2 (синдром Ди Джорджи / велокардиофациальный синдром)». Медицина (Балтимор). 90 (1): 1–18. Дои:10.1097 / MD.0b013e3182060469. PMID 21200182.
- ^ Деббане М., Глейзер Б., Дэвид М.К., Файнштейн С., Элиез С. (2006). «Психотические симптомы у детей и подростков с синдромом делеции 22q11.2: нейропсихологические и поведенческие последствия». Schizophr. Res. 84 (2–3): 187–93. Дои:10.1016 / j.schres.2006.01.019. PMID 16545541.
- ^ [неосновной источник необходим ] Бассетт А.С., Чоу Э.В., АбдельМалик П., Георгиу М., Хустед Дж., Вексберг Р. (2003). «Фенотип шизофрении при синдроме делеции 22q11». Am J Psychiatry. 160 (9): 1580–6. Дои:10.1176 / appi.ajp.160.9.1580. ЧВК 3276594. PMID 12944331.
- ^ [неосновной источник необходим ] Горовиц А., Шифман С., Ривлин Н., Пизанте А., Дарваси А. (2005). «Исследование микроделеции 22q11 в большой когорте пациентов с шизофренией». Schizophr. Res. 73 (2–3): 263–7. Дои:10.1016 / j.schres.2004.02.008. PMID 15653270.
- ^ Burn J (октябрь 1999 г.). «Время закрытия CATCH22». J. Med. Genet. 36 (10): 737–8. Дои:10.1136 / jmg.36.10.737. ЧВК 1734243. PMID 10528851.
- ^ а б Линдси EA (ноябрь 2001 г.). «Хромосомные микроделеции: расслаивающий синдром del22q11». Nat. Преподобный Жене. 2 (11): 858–68. Дои:10.1038/35098574. PMID 11715041.
- ^ Суиллен А., Фогельс А., Девриендт К., Фринс Дж. П. (2000). «Синдром делеции хромосомы 22q11: обновление и обзор клинических особенностей, когнитивно-поведенческого спектра и психиатрических осложнений». Являюсь. J. Med. Genet. 97 (2): 128–35. Дои:10.1002 / 1096-8628 (200022) 97: 2 <128 :: AID-AJMG4> 3.0.CO; 2-Z. PMID 11180220.
- ^ Малдун М., Оусли О.Ю., Кобрински Л.Дж., Патель С., Остер М.Э., Фернандес-Карриба С., Кубеллс Дж. Ф., Коулман К., Пирс Б.Д. (сентябрь 2015 г.). «Влияние гипокальциемии в раннем детстве на связанные с аутизмом социальные и коммуникативные навыки у пациентов с синдромом делеции 22q11». Eur Arch Psychiatry Clin Neurosci. 265 (6): 519–24. Дои:10.1007 / s00406-014-0546-0. ЧВК 4379129. PMID 25267002.
- ^ Цинксток Дж, ван Амельсвоорт Т (2005). «Нейропсихологический профиль и нейровизуализация у пациентов с синдромом делеции 22Q11.2: обзор». Детский нейропсихол. 11 (1): 21–37. Дои:10.1080/09297040590911194. PMID 15823981.
- ^ а б Мясник Н.Дж., Кил Т.Р., Хазрати Л.Н., Чоу Е.В., Рогаева Е., Ланг А.Е., Бассет А.С. (2013). «Связь между ранним началом болезни Паркинсона и синдромом делеции 22q11.2: идентификация новой генетической формы болезни Паркинсона и ее клинических последствий». JAMA Neurol. 70 (11): 1359–66. Дои:10.1001 / jamaneurol.2013.3646. ЧВК 4464823. PMID 24018986.
- ^ Мок К.Ю., Ширин Ю., Симон-Санчес Дж., Салака А., Честер Л., Эскотт-Прайс В. и др. (Май 2016). «Делеции 22q11.2 при идиопатической болезни Паркинсона: комбинированный анализ данных ассоциации по всему геному». Ланцет Нейрол. 15 (6): 585–96. Дои:10.1016 / S1474-4422 (16) 00071-5. ЧВК 4828586. PMID 27017469.
- ^ а б c Д'Антонио LL, Шерер NJ, Миллер LL, Kalbfleisch JH, Бартли JA (2001). «Анализ характеристик речи у детей с велокардиофациальным синдромом (VCFS) и детей с фенотипическим перекрытием без VCFS». Краниофак волчьей пасти. J. 38 (5): 455–67. Дои:10.1597 / 1545-1569 (2001) 038 <0455: AOSCIC> 2.0.CO; 2. ISSN 1545-1569. PMID 11522167.
- ^ а б c Шерер, штат Нью-Джерси, Д'Антонио, LL, Kalbfleisch JH (1999). «Раннее развитие речи и языка у детей с велокардиофациальным синдромом». Являюсь. J. Med. Genet. 88 (6): 714–23. Дои:10.1002 / (SICI) 1096-8628 (19991215) 88: 6 <714 :: AID-AJMG24> 3.0.CO; 2-B. PMID 10581495.
- ^ Шерер Нью-Джерси, Д'Антонио LL, Роджерс-младший (2001). «Профили коммуникативных расстройств у детей с велокардиофациальным синдромом: сравнение с детьми с синдромом Дауна». Genet. Med. 3 (1): 72–8. Дои:10.1097/00125817-200101000-00016. PMID 11339384.
- ^ Элиез С., Паласио-Эспаса Ф, Спира А (2000). «Маленькие дети с вело-кардио-лицевым синдромом (CATCH-22). Психологические и языковые фенотипы». Eur Детская подростковая психиатрия. 9 (2): 109–14. Дои:10.1007 / s007870050005. PMID 10926060.
- ^ а б c Робин Н.Х., Шпринцен Р.Дж. (2005). «Определение клинического спектра делеции 22q11.2». J. Pediatr. 147 (1): 90–6. Дои:10.1016 / j.jpeds.2005.03.007. PMID 16027702.
- ^ а б Солот CB, Knightly C, Handler SD (2000). «Коммуникационные расстройства при синдроме микроделеции 22Q11.2». J Commun Disord. 33 (3): 187–203, викторина 203–4. Дои:10.1016 / S0021-9924 (00) 00018-6. PMID 10907715.
- ^ Перссон С., Никлассон Л., Оскарсдоттир С., Йоханссон С., Йёнссон Р., Сёдерпалм Э (2006). «Языковые навыки у детей 5-8 лет с синдромом делеции 22q11». Int J Lang Commun Disord. 41 (3): 313–33. Дои:10.1080/13682820500361497. PMID 16702096.
- ^ а б c Онлайн-менделевское наследование в человеке (OMIM): #188400
- ^ а б Packham EA, Brook JD (апрель 2003 г.). «Гены Т-бокса при заболеваниях человека». Гм. Мол. Genet. 12 Спец. № 1 (90001): Р37–44. Дои:10.1093 / hmg / ddg077. PMID 12668595.
- ^ Тан К.Л., Антшел К.М., Фремонт В.П., Кейтс В.Р. (октябрь 2015 г.). «Поведенческие и психиатрические фенотипы при синдроме делеции 22q11.2». J Dev Behav Pediatr. 36 (8): 639–50. Дои:10.1097 / DBP.0000000000000210. ЧВК 4586411. PMID 26372046.
- ^ Maynard TM, Meechan DW, Dudevoir ML, Gopalakrishna D, Peters AZ, Heindel CC, Sugimoto TJ, Wu Y, Lieberman JA, Lamantia AS (ноябрь 2008 г.). «Митохондриальная локализация и функция подмножества генов-кандидатов синдрома делеции 22q11». Мол. Клетка. Неврологи. 39 (3): 439–51. Дои:10.1016 / j.mcn.2008.07.027. ЧВК 2729512. PMID 18775783.
- ^ а б Bartsch O, Nemecková M, Kocárek E, Wagner A, Puchmajerová A, Poppe M, Ounap K, Goetz P (февраль 2003 г.). «ДиДжордж / велокардиофациальный синдром: исследования FISH хромосом 22q11 и 10p14, а также клинические отчеты о проксимальной делеции 22q11». Являюсь. J. Med. Genet. А. 117A (1): 1–5. Дои:10.1002 / ajmg.a.10914. PMID 12548732.
- ^ Пуарсье С., Бессо-Аяссе Дж., Шлут-Болар С., Тутен Дж., Миссириан К., Ле Кайнек С. и др. (Июнь 2016). «Французское многоцентровое исследование более 700 пациентов с делециями 22q11, диагностированными с помощью FISH или aCGH». Евро. J. Hum. Genet. 24 (6): 844–51. Дои:10.1038 / ejhg.2015.219. ЧВК 4867458. PMID 26508576.
- ^ Эдельманн Л., Пандита Р.К., Спитери Е., Функе Б., Голдберг Р., Паланисами Н., Чаганти Р.С., Магенис Е., Шпринцен Р.Дж., Морроу Б.Е. (1999). «Общая молекулярная основа нарушений перестройки хромосомы 22q11». Хум Мол Генет. 8 (7): 1157–67. Дои:10.1093 / hmg / 8.7.1157. PMID 10369860.
- ^ Джером Л.А., Папайоанну В.Е. (март 2001 г.). «Фенотип синдрома ДиДжорджи у мышей, мутантных по гену Т-бокса, Tbx1». Nat. Genet. 27 (3): 286–91. Дои:10.1038/85845. PMID 11242110.
- ^ Линдси Э.А., Вителли Ф., Су Х., Моришима М., Хюинх Т., Прампаро Т., Юречич В., Огунрину Дж., Сазерленд Х.Ф., Скамблер П.Дж., Брэдли А., Балдини А. (март 2001 г.). «Tbx1 haploinsufficieny в области синдрома ДиДжорджи вызывает дефекты дуги аорты у мышей». Природа. 410 (6824): 97–101. Дои:10.1038/35065105. PMID 11242049.
- ^ Chun S, Du F, Westmoreland JJ, Han SB, Wang YD, Eddins D, et al. (Январь 2017 г.). «Таламический miR-338-3p опосредует слуховое таламокортикальное нарушение и его позднее начало в моделях микроделеции 22q11.2». Nat. Med. 23 (1): 39–48. Дои:10,1038 / нм.4240. ЧВК 5218899. PMID 27892953.
- ^ «Джин (NCBI)».
- ^ а б Кремер Л.С., Дистельмайер Ф., Альхаддад Б., Хемпель М., Юсо А., Кюппер С. и др. (2016). «Биаллельные усекающие мутации в TANGO2 вызывают рецидивирующие метаболические кризисы в младенчестве с энцефалокардиомиопатией». Американский журнал генетики человека. 98 (2): 358–62. Дои:10.1016 / j.ajhg.2015.12.009. ЧВК 4746337. PMID 26805782.
- ^ а б Лалани С.Р., Лю П., Розенфельд Дж. А., Уоткин Л. Б., Чан Ти, Ледук М.С. и др. (2016). «Рецидивирующая мышечная слабость с рабдомиолизом, метаболическими кризисами и сердечной аритмией из-за биаллельных мутаций TANGO2». Американский журнал генетики человека. 98 (2): 347–57. Дои:10.1016 / j.ajhg.2015.12.008. ЧВК 4746334. PMID 26805781.
- ^ Тонелли А.Р., Косури К., Вей С., Цыпленок Д. (2007). «Судороги как первое проявление синдрома делеции хромосомы 22q11.2 у 40-летнего мужчины: история болезни». Представитель J Med. 1: 167. Дои:10.1186/1752-1947-1-167. ЧВК 2222674. PMID 18053182.
- ^ Миллер, Кимберли А. (2008). «Диагностика FISH синдрома делеции 22q11.2». Отзывы о кормлении новорожденных и младенцев. 8 (1): e11 – e19. Дои:10.1053 / j.nainr.2007.12.006.
- ^ Джалали Г.Р., Ворстман Дж. А., Эррами А., Виджелаар Р., Бигель Дж., Шейх Т., Эмануэль Б. С. (март 2008 г.). «Детальный анализ 22q11.2 с помощью набора зондов высокой плотности MLPA». Гм. Мутат. 29 (3): 433–40. Дои:10.1002 / humu.20640. ЧВК 2664158. PMID 18033723.
- ^ Гарсия-Эрреро С., Кампос-Галиндо I, Мартинес-Конехеро Х.А., Серра V, Ольмо I, Лара С., Симон С., Рубио С. (2014). «Технология BACs-on-Beads: надежный тест для быстрого обнаружения анеуплоидий и микроделеций в пренатальной диагностике». Биомед Рес Инт. 2014: 590298. Дои:10.1155/2014/590298. ЧВК 3985206. PMID 24795887.
- ^ Choy KW, Kwok YK, Cheng YK, Wong KM, Wong HK, Leung KO, Suen KW, Adler K, Wang CC, Lau TK, Schermer MJ, Lao TT, Leung TY (сентябрь 2014 г.). «Диагностическая точность анализа BACs-on-Beads ™ по сравнению с кариотипированием для пренатального обнаружения хромосомных аномалий: ретроспективная серия последовательных случаев». BJOG. 121 (10): 1245–52. Дои:10.1111/1471-0528.12873. PMID 24893808.
- ^ Пак С.Дж., Чон Э.Х., Рю Р.С., Кан Х.В., Ко Дж.М., Ким Х.Д., Чхон С.К., Хван Ш., Кан Х.Й. (май 2011 г.). «Клиническое внедрение полногеномного массива CGH в качестве теста первого уровня в 5080 пре- и постнатальных случаях». Мол Цитогенет. 4: 12. Дои:10.1186/1755-8166-4-12. ЧВК 3114015. PMID 21549014.
- ^ Mupanemunda, Richard H .; Уоткинсон, Майкл (2004). Ключевые темы неонатологии. CRC Press. п. 82. ISBN 9781859962343.
- ^ «Синдром ДиДжорджи (синдром делеции 22q11.2)». Клиника Майо. Получено 22 мая 2020.
- ^ «Синдром ДиДжорджи (делеция 22q11.2): лечение и прогноз». www.uptodate.com. Получено 2018-10-30.
- ^ Markert ML, Devlin BH, Alexieff MJ, Li J, McCarthy EA, Gupton SE и др. (Май 2007 г.). «Обзор 54 пациентов с полной аномалией ДиДжорджи, включенных в протоколы трансплантации тимуса: результаты 44 последовательных трансплантаций». Кровь. 109 (10): 4539–47. Дои:10.1182 / кровь-2006-10-048652. ЧВК 1885498. PMID 17284531.
- ^ "Клиническая и метаболическая генетика - Клиника удаления 22q". Больница для больных детей. В архиве из оригинала от 07.04.2016.
- ^ Фунг В.Л., Мясник Н.Дж., Костейн Дж., Андраде Д.М., Бут E, Чоу Е.В. и др. (Август 2015 г.). «Практические рекомендации по ведению взрослых с синдромом делеции 22q11.2». Genet. Med. 17 (8): 599–609. Дои:10.1038 / гим.2014.175. ЧВК 4526275. PMID 25569435.
- ^ Оскарсдоттир С., Вуйч М., Фаст А. (2004). «Заболеваемость и распространенность синдрома делеции 22q11: популяционное исследование в Западной Швеции». Arch. Dis. Ребенок. 89 (2): 148–51. Дои:10.1136 / adc.2003.026880. ЧВК 1719787. PMID 14736631.
- ^ Daily DK, Ardinger HH, Holmes GE (февраль 2000 г.). «Выявление и оценка умственной отсталости». Am Fam Врач. 61 (4): 1059–67, 1070. PMID 10706158.
- ^ «Генетика 22q11.2 DS: Демография». Информация для медицинских работников. Dalglish Family Hearts and Minds Clinic для взрослых с синдромом делеции 22q11.2. В архиве из оригинала 9 марта 2016 г.. Получено 26 августа 2015.
- ^ «Синдром Ди Джорджа». Код диагноза D82.1 по МКБ-10-CM 2015 г.. В архиве из оригинала 24 сентября 2015 г.. Получено 26 августа 2015.
- ^ а б «Велокардио-лицевой синдром». Код диагноза Q93.81 по МКБ-10-CM 2015 г.. В архиве из оригинала 24 сентября 2015 г.. Получено 26 августа 2015.
- ^ Bassett AS, McDonald-McGinn DM, Devriendt K, Digilio MC, Goldenberg P, Habel A, Marino B, Oskarsdottir S, Philip N, Sullivan K, Swillen A, Vorstman J (август 2011 г.). «Практические рекомендации по ведению пациентов с синдромом делеции 22q11.2». J. Pediatr. 159 (2): 332–9.e1. Дои:10.1016 / j.jpeds.2011.02.039. ЧВК 3197829. PMID 21570089.
- ^ "Кампания с одинаковым именем - 22q.org". 22q.org. В архиве с оригинала на 2017-06-10. Получено 2017-06-18.
Эта статья включает текст из общественного достояния из Национальная медицинская библиотека США
внешняя ссылка
| Классификация | |
|---|---|
| Внешние ресурсы |
- Синдром ДиДжорджи в Керли
- McDonald-McGinn DM, Emanuel BS, Zackai EH (16 декабря 2005 г.). «Синдром делеции 22q11.2». В Pagon RA, Bird TD, Dolan CR, Stephens K (ред.). GeneReviews. PMID 20301696. NBK1523.
- Firth HV (17 февраля 2009 г.). «22q11.2 Дублирование». В Pagon RA, Bird TD, Dolan CR, Stephens K (ред.). GeneReviews. PMID 20301749. NBK3823.
| Основные темы | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Подходы | |||||||||||
| Права, закон, поддержка |
| ||||||||||
| Структурные и вспомогательные | |||||||||||
| Социальные проблемы | |||||||||||
| Искусство, СМИ, культура, спорт | |||||||||||
| |||||||||||