Болезнь Хантингтона - Huntingtons disease - Wikipedia
Эта статья должна быть обновлено. (Март 2020 г.) |
| болезнь Хантингтона | |
|---|---|
| Другие имена | Хорея Хантингтона |
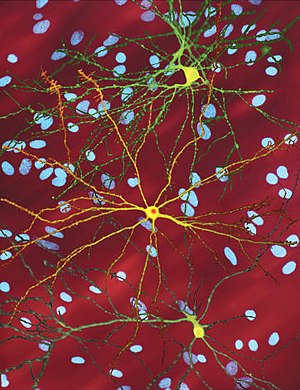 | |
| Отредактированное микроскопическое изображение средний колючий нейрон (желтый) с тело включения (оранжевый), который возникает как часть процесса болезни (ширина изображения 360мкм ) | |
| Специальность | Неврология |
| Симптомы | Проблемы с моторикой, включая координацию и походку, настроение и умственные способности.[1][2] |
| Осложнения | Пневмония, сердечное заболевание, телесные повреждения в результате падений, самоубийство[3] |
| Обычное начало | 30–50 лет[4] |
| Продолжительность | Долгосрочный[4] |
| Причины | Генетический (унаследованная или новая мутация)[4] |
| Диагностический метод | Генетическое тестирование[5] |
| Дифференциальная диагностика | Хорея Сиденхема, доброкачественная наследственная хорея, волчанка, паранеопластический синдром, Болезнь Вильсона[6] |
| Уход | Поддерживающая терапия[2] |
| Медикамент | Тетрабеназин[3] |
| Прогноз | 15–20 лет с момента постановки диагноза[4] |
| Частота | 4–15 из 100 000 (европейское происхождение)[1] |
болезнь Хантингтона (HD), также известный как Хорея Хантингтона, это нейродегенеративное заболевание это в основном унаследованный.[7] Самые ранние симптомы - это часто незаметные проблемы с настроением или умственными способностями.[1] Генерал отсутствие координации и неустойчивый походка часто следуют.[2] По мере развития болезни нескоординированные непроизвольные движения тела, известные как хорея становятся более очевидными.[1] Физические способности постепенно ухудшаются, пока согласованное движение становится трудно, и человек не может говорить.[1][2] Умственные способности обычно снижается до слабоумие.[3] Специфические симптомы у разных людей несколько различаются.[1] Симптомы обычно появляются в возрасте от 30 до 50 лет, но могут появиться в любом возрасте.[4][3] Заболевание может развиваться раньше в каждом следующем поколении.[1] Около восьми процентов случаев начинаются в возрасте до 20 лет и известны как несовершеннолетний HD, которые обычно присутствуют с симптомы замедленного движения из болезнь Паркинсона а не хореи.[3]
HD обычно унаследован от пострадавшего родителя, который носит мутация в ген хантингтина (HTT).[4] Однако до 10% случаев связаны с новой мутацией.[1] Ген хантингтина предоставляет генетическую информацию для белок хантингтин (htt).[1] Расширение CAG повторяется из цитозин -аденин -гуанин (известный как экспансия тринуклеотидного повтора ) в гене, кодирующем белок хантингтина, приводит к аномальному мутантному белку (mhtt), который постепенно повреждает клетки мозга с помощью ряда возможных механизмов.[7][8] Диагноз ставится генетическое тестирование, которые можно проводить в любое время, независимо от наличия симптомов.[5] Этот факт вызывает несколько этических споров: возраст, в котором человек считается достаточно зрелым, чтобы выбрать тестирование; имеют ли родители право на тестирование своих детей; и управление конфиденциальностью и раскрытием результатов тестирования.[2]
Лекарства от HD не существует, и на более поздних стадиях требуется постоянный уход.[2] Лечение может облегчить некоторые симптомы, а в некоторых - улучшить качество жизни.[3] Лучшее доказательство для лечения проблем с движением - это тетрабеназин.[3] HD поражает от 4 до 15 человек европейского происхождения на 100 000 человек.[1][3] Это редкость среди японцев, а частота встречаемости в Африке неизвестна.[3] Заболевание одинаково поражает мужчин и женщин.[3] Такие осложнения, как пневмония, сердечное заболевание, и телесные повреждения в результате падений сокращают продолжительность жизни.[3] Самоубийство является причиной смерти примерно в 9% случаев.[3] Смерть обычно наступает через 15–20 лет после того, как болезнь была впервые обнаружена.[4]
Первое вероятное описание болезни было в 1841 году американским врачом Чарльзом Оскаром Уотерсом.[9] Более подробно это состояние было описано в 1872 году американским врачом. Джордж Хантингтон.[9] Генетическая основа была открыта в 1993 году совместными международными усилиями под руководством Фонд наследственных болезней.[10][11] Исследования и вспомогательные организации начал формироваться в конце 1960-х годов с целью повышения осведомленности общественности, оказания поддержки отдельным лицам и их семьям и содействия исследованиям.[12][11] Направления исследований включают определение точного механизма заболевания, улучшение животные модели для помощи в исследованиях, тестировании лекарств для лечения симптомов или замедления прогрессирования заболевания, а также в изучении таких процедур, как терапия стволовыми клетками с целью замены поврежденных или потерянных нейронов.[10]
Признаки и симптомы
| Раздражительность | 38–73% |
| Апатия | 34–76% |
| Беспокойство | 34–61% |
| В депрессии | 33–69% |
| Обсессивно-компульсивный | 10–52% |
| Психотический | 3–11% |
Симптомы болезни Гентингтона чаще всего становятся заметными в возрасте от 30 до 50 лет, но они могут начаться в любом возрасте.[4] Их прогрессирование часто описывается на ранних, средних и поздних стадиях с более ранней продромальной фазой.[2] На ранних стадиях наблюдаются тонкие изменения личности, проблемы в познание, а также физические навыки, раздражительность и перепады настроения - все это может остаться незамеченным,[14][15] и они обычно предшествуют двигательным симптомам.[16] Почти у всех с HD в конечном итоге проявляются схожие физические симптомы, но начало, прогрессирование и степень когнитивных и поведенческих симптомов значительно различаются у разных людей.[17][18]
Наиболее характерными начальными физическими симптомами являются резкие, случайные и неконтролируемые движения, называемые хорея.[19] Многие люди не осознают своих непроизвольных движений или им мешают.[1] Первоначально хорея может проявляться как общее беспокойство, небольшие непреднамеренно инициированные или незавершенные движения, отсутствие координации или замедление. саккадические движения глаз.[19] Эти незначительные двигательные нарушения обычно предшествуют более очевидным признакам двигательной дисфункции как минимум на три года.[17] Явное появление таких симптомов, как ригидность, корчащиеся движения или ненормальная поза появляются по мере прогрессирования расстройства.[19] Это признаки того, что система мозга, отвечающая за движение, была затронута.[20] Психомоторный функции становятся все более и более нарушенными, так что любое действие, требующее мышечного контроля, нарушается. Общие последствия - физическая нестабильность, неправильное выражение лица и трудности с жеванием. глотание, и Говорящий.[19] Нарушения сна и потеря веса также сопутствующие симптомы.[21] Проблемы с питанием обычно вызывают потерю веса и могут привести к недоеданию.[22][23] Ювенильная HD обычно прогрессирует более быстрыми темпами, быстрее с большим когнитивным снижением, и хорея проявляется ненадолго, если вообще проявляется; то Вестфальский вариант из замедленность движения, ригидность и тремор более характерны для ювенильного HD, как и припадки.[19][21]
Познавательные способности постепенно ухудшаются.[20] Особенно страдают исполнительные функции, которые включают планирование, когнитивную гибкость, абстрактное мышление, получение правил, инициирование соответствующих действий и запрещение несоответствующих действий.[20] По мере прогрессирования болезни объем памяти появляются дефициты. Зарегистрированные нарушения варьируются от краткосрочная память дефицит Долгосрочная память трудности, в том числе дефицит эпизодический (память о своей жизни), процедурный (память тела о том, как выполнять действие) и рабочая память.[20] Когнитивные проблемы со временем усугубляются, что в конечном итоге приводит к слабоумие.[20]
Сообщается психоневрологический знаки беспокойство, депрессия, а снижение проявления эмоций, эгоцентризм, агрессия, и компульсивное поведение, последнее из которых может вызвать или ухудшить пристрастия, включая алкоголизм, играть в азартные игры, и гиперсексуальность.[13] Также наблюдались трудности с распознаванием негативных выражений других людей.[20] В распространенность этих симптомов сильно различается между исследованиями, с оценкой распространенности в течение жизни психические расстройства от 33% до 76%.[13] Для многих больных и их семей эти симптомы являются одними из самых тревожных аспектов болезни, часто влияя на повседневное функционирование и являясь причиной институционализация.[13] Суицидальные мысли и попытки самоубийства встречаются чаще, чем среди населения в целом.[19] Часто люди не осознают хорею, когнитивные и эмоциональные нарушения.[24]
Мутантный хантингтин экспрессируется по всему телу и связан с аномалиями в периферических тканях, которые непосредственно вызваны такой экспрессией вне мозга. Эти отклонения включают: мышечная атрофия, сердечная недостаточность, нарушенной толерантности к глюкозе, потеря веса, остеопороз, и атрофия яичек.[25]
Генетика
У каждого есть две копии ген хантингтина (HTT), который кодирует белок хантингтин (htt). HTT также называют Ген HD, а Ген IT15, (интересно стенограмма 15). Часть этого гена представляет собой повторяющийся участок, называемый экспансия тринуклеотидного повтора - а короткий повтор длина которого варьируется от человека к человеку и может меняться от поколения к поколению. Если повтор присутствует в здоровом гене, динамическая мутация может увеличить количество повторов и привести к дефектному гену. Когда длина этого повторяющегося участка достигает определенного порога, он производит измененную форму белка, называемую мутантным белком хантингтина (mhtt). Различные функции этих белков являются причиной патологических изменений, которые, в свою очередь, вызывают симптомы болезни. Мутация при болезни Хантингтона является генетически доминирующей и почти полностью пенетрант: мутация любого из HTT аллели вызывают заболевание. Он наследуется не по полу, а по длине повторяющегося участка гена, и, следовательно, его серьезность может зависеть от пола пораженного родителя.[19]
Генетическая мутация
HD - один из нескольких нарушения тринуклеотидного повтора которые вызваны превышением длины повторяющегося участка гена нормального диапазона.[19] В HTT ген находится на короткая рука из хромосома 4[19] при 4п16.3. HTT содержит последовательность из трех Основания ДНК - цитозин-аденин-гуанин (CAG) - повторяется несколько раз (т.е. ... CAGCAGCAG ...), известный как тринуклеотидный повтор.[19] CAG - трехбуквенный генетический код (кодон ) для аминокислота глутамин, поэтому ряд из них приводит к производству цепочки глютамина, известной как полиглутаминовый тракт (или polyQ тракт), и повторяющаяся часть гена, Регион PolyQ.[26]
| Количество повторов | Классификация | Статус болезни | Риск для потомства |
|---|---|---|---|
| <27 | Нормальный | Не пострадает | Никто |
| 27–35 | Средний | Не пострадает | Повышенный, но <50% |
| 36–39 | Пониженная проницаемость | Может или не может быть затронут | 50% |
| 40+ | Полное проникновение | Будет затронуто | 50% |
Как правило, люди имеют менее 36 повторяющихся глутаминов в области polyQ, что приводит к выработке цитоплазматический протеин хантингтин.[19] Однако последовательность из 36 или более глутаминов приводит к продукции белка, который имеет разные характеристики.[19] Эта измененная форма, называемая мутантным хантингтином (mhtt), увеличивает скорость распада некоторых типов нейроны. Области мозга имеют разное количество и зависят от этих типов нейронов, и на них влияют соответственно.[19] Как правило, количество повторов CAG зависит от того, насколько сильно затронут этот процесс, и составляет около 60% вариации возраста появления симптомов. Оставшаяся вариация связана с окружающей средой и другими генами, которые модифицируют механизм HD.[19] 36–39 повторов приводят к форме болезни со сниженной пенетрантностью, с гораздо более поздним началом и более медленным прогрессированием симптомов. В некоторых случаях начало может быть настолько поздним, что симптомы даже не заметны.[19] При очень большом количестве повторений (более 60) начало HD может произойти в возрасте до 20 лет, известное как несовершеннолетний HD. Ювенильный HD обычно Вестфальский вариант который характеризуется замедленностью движений, ригидностью и тремором. Это около 7% носителей HD.[27][28]
Наследование
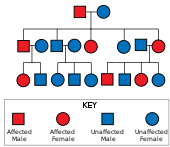
Болезнь Хантингтона имеет аутосомно-доминантный наследование, означающее, что пораженный человек обычно наследует одну копию гена с расширенным тринуклеотидным повторением (мутантный аллель ) от пострадавшего родителя.[19] Поскольку пенетрантность мутации очень высока, те, у кого есть мутированная копия гена, будут болеть. При этом типе наследования каждое потомство пораженного индивидуума имеет 50% -ный риск наследования мутантного аллеля и, следовательно, поражение этим заболеванием (см. Рисунок). Эта вероятность не зависит от пола.[29]
Тринуклеотидные CAG-повторы более 28 лет нестабильны во время репликация, и эта нестабильность увеличивается с увеличением количества присутствующих повторов.[19] Обычно это приводит к новым расширениям по мере смены поколений (динамические мутации ) вместо воспроизведения точной копии тринуклеотидного повтора.[19] Это вызывает изменение количества повторов в последовательных поколениях, так что здоровый родитель с «промежуточным» количеством повторов (28–35) или «пониженной пенетрантностью» (36–40) может передать копию гена. с увеличением числа повторов, что дает полностью пенетрантный HD.[19] Такое увеличение количества повторов (а значит, и более раннее возраст начала и тяжесть заболевания) в следующих поколениях называется генетическим ожидание.[1] Нестабильность больше в сперматогенез чем оогенез;[19] Унаследованные от матери аллели обычно имеют одинаковую длину повтора, тогда как аллели, унаследованные от отца, имеют больше шансов на увеличение длины.[19][30] Болезнь Хантингтона редко бывает вызвана новая мутация, где ни один из родителей не имеет более 36 повторов CAG.[31]
В редких ситуациях, когда оба родителя имеют расширенный ген HD, риск увеличивается до 75%, а когда у любого из родителей есть две расширенные копии, риск составляет 100% (затронуты все дети). Лица с оба гена затронуты редки. Некоторое время считалось, что HD является единственным заболеванием, при котором наличие второго мутированного гена не влияет на симптомы и прогрессирование.[32] но с тех пор было обнаружено, что это может повлиять на фенотип и скорость прогрессирования.[19][33]
Механизмы
Белок хантингтин взаимодействует с более чем 100 другими белками и, по-видимому, выполняет несколько функций.[34] Поведение мутировавшего белка (mhtt) до конца не изучено, но он токсичен для определенных типов клеток, особенно в головном мозге. Раннее повреждение наиболее очевидно в полосатое тело, но по мере прогрессирования заболевания другие области мозга также более заметно поражаются. Ранние симптомы связаны с функциями полосатого тела и его корковых связей, а именно с контролем над движением, настроением и высшими когнитивными функциями.[19] Метилирование ДНК тоже кажется измененным в HD.[35]
Функция Хантингтина
Хантингтин (HTT) - это выразил во всех клетках, с самыми высокими концентрациями в головном мозге и яички, и умеренные количества в печень, сердце, и легкие. Однако его функция неясна.[19] Он взаимодействует с белками, которые участвуют в транскрипции, клеточная сигнализация, и внутриклеточные транспортировка.[19][36] У животных генетически модифицированный для проявления HD были идентифицированы несколько функций HTT.[37] У этих животных HTT важна для эмбрионального развития, поскольку ее отсутствие связано с гибелью эмбриона. Caspase, фермент, который играет роль в катализе апоптоза, как полагают, активируется мутировавшим геном через повреждение системы убиквитин-протеаза. Он также действует как антиапоптотический средство предотвращения запрограммированная гибель клеток и контролирует производство нейротрофический фактор головного мозга, белок, который защищает нейроны и регулирует их создание во время нейрогенез. HTT также способствует везикулярный транспорт и синаптическая передача и контролирует транскрипцию нейрональных генов.[37] Если выражение HTT увеличивается и производится больше HTT, клетка мозга выживаемость улучшается, а эффекты mhtt снижаются, тогда как когда экспрессия HTT снижается, результирующие характеристики становятся более заметными в присутствии mhtt.[37] Соответственно считается, что болезнь вызвана не неадекватное производство HTT, но токсическое усиление функции МХТТ в теле.[19]
Сотовые изменения
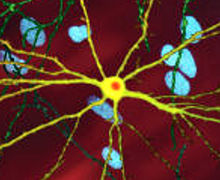
Существует множество клеточных изменений, через которые может проявляться токсическое действие mhtt и приводить к патологии HD.[38][39] В своей мутантной (т. Е. Расширенной полиглутамином) форме белок более склонен к расщеплению, которое создает более короткие фрагменты, содержащие расширение полиглутамина.[38] Эти белковые фрагменты склонны к неправильная укладка и агрегация, дающая фибриллярные агрегаты, в которых ненативные β-цепи полиглутамина из множества белков связаны вместе посредством водородных связей.[8] Эти агрегаты имеют общий фундаментальный кросс-β амилоид архитектура наблюдается при других заболеваниях отложений белка. Со временем агрегаты накапливаются, образуя органы включения внутри клеток, что в конечном итоге мешает работе нейронов.[38][8] Включения нейронов вызывают косвенное вмешательство. Тельца включения были обнаружены как в ядро клетки и цитоплазма.[38] Тельца включения в клетках головного мозга являются одним из самых ранних патологических изменений, и некоторые эксперименты показали, что они могут быть токсичный для клетки, но другие эксперименты показали, что они могут формироваться как часть защитного механизма организма и помогать защищать клетки.[38]
Идентифицировано несколько путей, по которым mhtt может вызывать гибель клеток. К ним относятся: влияние на белки-шапероны, которые помогают сворачивать белки и удалять неправильно свернутые; взаимодействие с каспасы, которые играют роль в процесс удаления клеток; то токсическое действие глутамина на нервные клетки; нарушение выработки энергии внутри клеток; и влияние на экспрессию генов.[8][40]
Мутант Хантингтин было обнаружено, что белок играет ключевую роль в митохондриальный дисфункция.[41] Нарушение митохондриальной электронный транспорт может привести к более высокому уровню окислительный стресс и выпуск активные формы кислорода.[42]
Глютамин известен как эксайтотоксический когда присутствуют в больших количествах, эксайтотоксины вызывают повреждение многочисленных клеточных структур. Глутамин не обнаруживается в чрезмерно высоких количествах при HD, но взаимодействия измененного белка хантингтина с многочисленными белками в нейронах приводят к повышенной уязвимости к глутамину. Было высказано предположение, что повышенная уязвимость приводит к эксайтотоксическим эффектам от нормального уровня глутамина.[8]
Макроскопические изменения

HD влияет на весь мозг, но некоторые области более уязвимы, чем другие. Наиболее заметные ранние эффекты находятся в части базальный ганглий называется полосатое тело, который состоит из хвостатое ядро и скорлупа.[19] Другие затронутые области включают черная субстанция, корковые слои 3, 5 и 6 из неокортекс, то гиппокамп, Клетки Пуркинье в мозжечок латеральные туберальные ядра гипоталамус и части таламус.[19] Эти области поражаются в соответствии с их структурой и типами нейронов, которые они содержат, уменьшаясь в размере по мере потери клеток.[19] Полосатый средние шиповатые нейроны являются наиболее уязвимыми, особенно с прогнозы навстречу внешний бледный шар, с интернейроны и колючие клетки, выступающие в внутренний бледный шар меньше подвержен влиянию.[19][43] HD также вызывает аномальное увеличение в астроциты и активация иммунных клеток головного мозга, микроглия.[44]
Базальные ганглии - часть мозга, наиболее сильно пораженная на ранних стадиях HD, - играют ключевую роль в контроле движений и поведения. Их функции до конца не изучены, но современные теории предполагают, что они являются частью когнитивного исполнительная система[20] и цепь двигателя.[45] Базальные ганглии обычно подавляют большое количество цепей, генерирующих определенные движения. Чтобы инициировать определенное движение, кора головного мозга посылает сигнал в базальные ганглии, который вызывает снятие торможения. Повреждение базальных ганглиев может вызвать беспорядочное и неконтролируемое снятие или восстановление запретов, что приводит к неловкому началу движения или движениям, которые могут быть инициированы непреднамеренно, или к остановке движения до или после предполагаемого завершения. Накапливающиеся повреждения этой области вызывают характерные беспорядочные движения, связанные с HD, известные как хорея, а дискинезия.[45] Из-за неспособности базальных ганглиев подавлять движения люди, затронутые им, неизбежно будут испытывать снижение способности говорить и глотать пищу и жидкости (дисфагия).[46]
Нарушение регуляции транскрипции
CREB-связывающий белок (CBP), корегулятор транскрипции, необходим для функционирования клеток, поскольку в качестве коактиватора значительного числа промоторов он активирует транскрипцию генов путей выживания.[40] Кроме того, аминокислоты, которые образуют CBP, включают полоску из 18 глутаминов. Таким образом, глутамины на CBP напрямую взаимодействуют с увеличенным количеством глутамина в цепи HTT, и CBP удаляется из своего типичного местоположения рядом с ядром.[47] В частности, CBP содержит домен ацетилтрансферазы, с которым HTT связывается через свой полиглутаминсодержащий домен.[48] Вскрытие мозга людей, перенесших болезнь Хантингтона, также показало невероятно низкое количество CBP.[47] Кроме того, когда CBP сверхэкспрессируется, гибель, вызванная полиглутамином, снижается, что дополнительно демонстрирует, что CBP играет важную роль в болезни Хантингтона и нейронов в целом.[40]
Диагностика
Медицинский диагноз Начало HD может быть установлено после появления физических симптомов, специфичных для данного заболевания.[19] Генетическое тестирование может использоваться для подтверждения физического диагноза, если в семейном анамнезе не было ГБ. Еще до появления симптомов генетическое тестирование может подтвердить, есть ли у человека или эмбрион несет расширенную копию тринуклеотидного повтора (CAG) в HTT ген, вызывающий заболевание. Генетическое консультирование доступен для предоставления рекомендаций и рекомендаций на протяжении всей процедуры тестирования, а также о последствиях подтвержденного диагноза. Эти последствия включают влияние на психологию человека, карьеру, решения в области планирования семьи, родственников и отношения. Несмотря на доступность предсимптоматического тестирования, только 5% из тех, кто имеет риск унаследовать HD, выбирают это.[19]
Клинический

А физический осмотр, иногда в сочетании с психологическое обследование, можно определить, началось ли начало болезни.[19] Чрезмерные непреднамеренные движения любой частью тела часто являются причиной обращения за медицинской помощью. Если они резкие, случайные по времени и распределению, они предполагают диагноз HD. Когнитивные или поведенческие симптомы редко являются первыми диагностированными симптомами; они обычно распознаются только задним числом или когда они развиваются дальше. Насколько далеко зашло заболевание, можно измерить с помощью унифицированная шкала оценки болезни Хантингтона, который обеспечивает общую систему оценок, основанную на моторных, поведенческих, когнитивных и функциональных оценках.[50][51] Медицинская визуализация, Такие как компьютерная томография (CT) и магнитно-резонансная томография (МРТ), может показать атрофию хвостатых ядер на ранней стадии заболевания, как показано на иллюстрации справа, но эти изменения сами по себе не являются диагностикой HD. Церебральная атрофия можно увидеть на запущенных стадиях болезни. Функциональная нейровизуализация методы, такие как функциональная магнитно-резонансная томография (фМРТ) и позитронно-эмиссионная томография (ПЭТ), могут показать изменения в активности мозга до появления физических симптомов, но они являются экспериментальными инструментами и не используются в клинических условиях.[19]
Прогностическое генетическое тестирование
Поскольку HD следует аутосомно-доминантному типу наследования, у людей, которые подвержены риску наследования, есть сильная мотивация искать диагноз. В генетический тест для HD состоит из анализ крови который подсчитывает количество повторов CAG в каждом из HTT аллели.[52] Отсечки представлены следующим образом:
- 40 и более CAG-повторов: полный пенетрантность аллель (FPA).[53] А "положительный тест "или" положительный результат "обычно относится к этому случаю. Положительный результат не считается диагнозом, поскольку он может быть получен за несколько десятилетий до появления симптомов. Однако отрицательный результат означает, что человек не несет расширенной копии гена. и не будет разрабатывать HD.[19] Тест скажет человеку, у которого изначально была 50-процентная вероятность унаследовать болезнь, если его риск возрастет до 100 процентов или будет устранен. У человека с положительным результатом теста на заболевание в какой-то момент разовьется HD, при условии, что он или она проживет достаточно долго, чтобы болезнь появилась.[19]
- От 36 до 39 повторов: неполный или же аллель пониженной пенетрантности (РПА). Это может вызвать симптомы, как правило, в более зрелом возрасте.[53] Существует максимальный риск 60% того, что у человека с RPA появятся симптомы в возрасте 65 лет, и 70% риск появления симптомов в возрасте 75 лет.[53]
- От 27 до 35 повторов: промежуточный аллель (IA), или большой нормальный аллель. Это не связано с симптоматическим заболеванием у тестируемого человека, но может расширяться при дальнейшем наследовании, давая симптомы у потомства.[53]
- 26 или меньше повторов: не связаны с HD.[53]
Тестирование до появления симптомов - событие, которое меняет жизнь, и это очень личное решение.[19] Основная причина выбора тестирования на HD - помощь в выборе карьеры и семьи.[19] До 1993 года не было доступного теста для людей, чтобы узнать, несут ли они ген Хантингтона. В то время опросы показали, что 50–70% людей из группы риска были бы заинтересованы в прохождении тестирования, но, поскольку было предложено прогностическое тестирование, гораздо меньшее число желающих пройти тестирование.[54] Более 95% людей с риском наследования БГ не проходят тестирование, в основном из-за отсутствия лечения.[19] Ключевой проблемой является тревога, которую человек испытывает по поводу незнания, разовьется ли у него в конечном итоге БГ, по сравнению с влиянием положительного результата.[19] Независимо от результата было установлено, что уровень стресса ниже через два года после тестирования, но риск самоубийства увеличивается после положительного результата теста.[19] Лица, у которых обнаружено не унаследованное заболевание, могут испытывать вина выжившего в отношении затронутых членов семьи.[19] Другие факторы, принимаемые во внимание при рассмотрении вопроса о тестировании, включают возможность дискриминации и последствия положительного результата, что обычно означает, что у одного из родителей есть пораженный ген, и что братья и сестры индивида рискуют унаследовать его.[19] В одном исследовании генетическая дискриминация была обнаружена у 46% лиц, подверженных риску болезни Хантингтона. Это происходило чаще в личных отношениях, чем при страховании здоровья или трудовых отношениях.[55] Генетическое консультирование in HD может предоставить информацию, советы и поддержку для первоначального принятия решений, а затем, если таковые выбраны, на всех этапах процесса тестирования.[56] Из-за последствий этого теста пациенты, которые хотят пройти тестирование, должны пройти три сеанса консультирования, которые предоставят информацию о болезни Хантингтона.[57]
Консультации и рекомендации по использованию генетического тестирования на HD стали моделями для других генетических заболеваний, таких как аутосомно-доминантное мозжечковая атаксия.[19][58][59] Пресимптоматическое тестирование для HD также повлиял на тестирование других заболеваний с генетическими вариантами, такими как поликистозная почка болезнь, семейная Болезнь Альцгеймера и рак молочной железы.[58] Европейская сеть качества молекулярной генетики опубликовала ежегодную схему внешней оценки качества для молекулярно-генетического тестирования на это заболевание и разработала рекомендации по передовой практике генетического тестирования на HD, чтобы помочь в тестировании и сообщении результатов.[60]
Преимплантационная генетическая диагностика
Эмбрионы произведено с использованием экстракорпоральное оплодотворение могут быть генетически протестированы на HD с использованием предимплантационная генетическая диагностика (ПГД). Этот метод, при котором одна или две клетки извлекаются из обычно 4-8-клеточного эмбриона и затем проверяются на генетическую аномалию, затем может использоваться, чтобы гарантировать, что эмбрионы, пораженные генами HD, не имплантированы, и, следовательно, любое потомство не будет наследовать болезнь. Некоторые формы доимплантационной генетической диагностики - тестирование на неразглашение или исключение - позволяют людям из группы риска иметь потомство без HD. без раскрытие собственного родительского генотипа, не давая информации о том, суждено ли им самим заболеть HD. При тестировании исключения ДНК эмбрионов сравнивается с ДНК родителей, бабушек и дедушек, чтобы избежать наследования хромосомной области, содержащей ген HD, от пораженной бабушки и дедушки. При тестировании на неразглашение информации в матке заменяются только здоровые эмбрионы, в то время как родительский генотип и, следовательно, родительский риск HD никогда не раскрываются.[61][62]
Пренатальное тестирование
Также возможно получить пренатальная диагностика для эмбриона или плод в утробе матери, используя генетический материал плода, полученный через биопсия хориона. An амниоцентез может быть выполнено при продолжении беременности в течение 14–18 недель. Эта процедура исследует околоплодные воды, окружающие ребенка, на предмет признаков мутации HD.[63] Это также можно сочетать с тестированием исключения, чтобы избежать раскрытия родительского генотипа. Пренатальное тестирование может быть выполнено, когда у родителя был диагностирован HD, когда у них было генетическое тестирование, показывающее расширение гена HTT, или когда у них есть 50% шанс унаследовать болезнь. Родители могут быть проконсультированы по поводу возможных вариантов, в том числе: прерывание беременности, и о трудностях ребенка с выявленным геном.[64][65]
Кроме того, при беременности с повышенным риском из-за пораженного партнера-мужчины неинвазивная пренатальная диагностика может быть выполнена путем анализа внеклеточная ДНК плода в пробе крови, взятой у матери (через венепункция ) между шестью и двенадцатью неделями беременности.[53] Отсутствует связанный с процедурой риск выкидыша.[53]
Дифференциальная диагностика
Около 99% диагнозов HD основаны на типичных симптомах и история семьи заболевания подтверждены генетическим тестированием на наличие увеличенного тринуклеотидного повтора, вызывающего HD. Большинство остальных называются HD-подобные (HDL) синдромы.[19][66] Причина большинства заболеваний ЛПВП неизвестна, но болезни с известными причинами связаны с мутациями в ген прионного белка (HDL1), ген юнктофилина 3 (HDL2), рецессивно наследуемый неизвестный ген (HDL3 - встречается только в двух семьях и плохо изучен), и ген, кодирующий ТАТА-бокс-связывающий белок (SCA17, иногда называемый HDL4 ). К другим аутосомно-доминантным заболеваниям, которые можно ошибочно принять за HD, относятся: дентаторубрально-паллидолуйзийская атрофия и нейроферритинопатия. Это также аутосомно-рецессивный расстройства, напоминающие спорадические случаи HD. К ним относятся хорея акантоцитоз и нейродегенерация, связанная с пантотенаткиназой. Один Х-связанный расстройство этого типа Синдром Маклеода.[66]
Управление
Лекарства от HD не существует, но существуют методы лечения, позволяющие уменьшить тяжесть некоторых из его симптомов.[67] Для многих из этих методов лечения доказательства, подтверждающие их эффективность в лечении симптомов HD, являются неполными.[19][68] По мере прогрессирования болезни способность заботиться о себе снижается, и ее следует тщательно контролировать. мультидисциплинарный забота становится все более необходимым.[19] Хотя исследований упражнений и методов лечения, которые помогают реабилитировать когнитивные симптомы БХ, есть некоторые доказательства полезности физиотерапия, трудотерапия, и Логопедия.[19]
Терапия
Похудание и проблемы с питанием из-за трудности с глотанием, и другие мышечные нарушения координации являются обычным явлением, что делает управление питанием все более важным по мере развития болезни.[19] Загустители можно добавлять в жидкости, так как более густые жидкости легче и безопаснее глотать.[19] Напоминание пострадавшему о том, что он должен есть медленно и брать в рот более мелкие кусочки пищи, также может быть полезным для предотвращения удушья.[19] Если еда становится слишком опасной или неудобной, можно использовать чрескожная эндоскопическая гастростомия доступен. Это питательная трубка, постоянно прикрепленная через брюшная полость в желудок, что снижает риск всасывающий питание и обеспечивает лучшее управление питанием.[69] Оценка и управление логопеды с опытом лечения болезни Гентингтона рекомендуется.[19]
Люди с болезнью Гентингтона могут видеть физиотерапевт для неинвазивных и немедикаментозных способов управления физическими симптомами. Физиотерапевты могут проводить оценку риска падений и предотвращать их, а также выполнять упражнения на укрепление, растяжку и сердечно-сосудистые упражнения. Вспомогательные средства для ходьбы могут быть прописаны при необходимости. Физиотерапевты также назначают дыхательные упражнения и методы очистки дыхательных путей при развитии респираторных проблем.[70] Консенсусное руководство по физиотерапии при болезни Хантингтона было разработано Европейская сеть HD.[70] Цели раннего реабилитация вмешательства - это предотвращение потери функции. Участие в программах реабилитации на ранней и средней стадии заболевания может быть полезным, так как способствует долгосрочному поддержанию двигательной и функциональной работоспособности. Реабилитация на поздней стадии направлена на компенсацию двигательных и функциональных потерь.[71] Для долгосрочного независимого ведения терапевт может разработать программы домашних упражнений для подходящих людей.[72]
Кроме того, все большее число людей с болезнью Хантингтона обращаются к паллиативной помощи, которая направлена на улучшение качества жизни за счет лечения симптомов и стресса тяжелого заболевания в дополнение к другим методам лечения.[73]
Лекарства
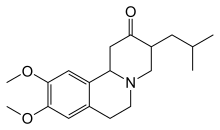
Тетрабеназин был одобрен в 2000 г. для лечения хореи при болезни Хантингтона в ЕС, а в 2008 г. - в США.[74] Другие препараты, которые помогают уменьшить хорею, включают: нейролептики и бензодиазепины.[15] Такие соединения как амантадин или же Ремасемид все еще расследуются, но уже показали предварительные положительные результаты.[19] Гипокинезия и ригидность, особенно у несовершеннолетних, можно лечить с помощью противопаркинсонический наркотики и миоклонический гиперкинез можно лечить с помощью вальпроевая кислота.[15] Предварительные доказательства найдены этил эйкозапентаеновая кислота для улучшения двигательных симптомов в один год.[75] В 2017 г. Дейетрабеназин Более тяжелая форма тетрабеназинового препарата для лечения хореи при HD была одобрена FDA.[76] Это продается как Остедо и это первый низкомолекулярный препарат получить одобрение FDA.[77]
Психиатрические симптомы можно лечить с помощью лекарств, аналогичных тем, которые используются в общей популяции.[19][68] Селективные ингибиторы обратного захвата серотонина и миртазапин рекомендованы при депрессии, а атипичные нейролептики рекомендуются для психоз и поведенческие проблемы.[68] Рекомендуется помощь специалиста психоневрологического профиля, поскольку людям может потребоваться длительное лечение с использованием комбинации нескольких препаратов.[19]
Образование
Семьи отдельных лиц и общество в целом, те, кто унаследовали или находятся в группе риска наследования HD, имеют опыт HD в течение нескольких поколений, но могут не знать о недавних достижениях в понимании этого заболевания и о доступности генетического тестирования. Генетическое консультирование приносит пользу этим людям, обновляя свои знания, стремясь развеять любые необоснованные убеждения, которые они могут иметь, и помогая им обдумать свои будущие варианты и планы. Также включена информация о вариантах планирования семьи, организации ухода и других аспектах.[19][78]
Прогноз
Длина тринуклеотидного повтора составляет 60% вариации возраста появления симптомов и скорости их развития. Более длительное повторение приводит к более раннему возрасту начала и более быстрому прогрессированию симптомов.[19][79] Люди с более чем шестидесяти повторениями часто заболевают до 20 лет, в то время как у лиц с менее чем 40 повторами симптомы могут отсутствовать.[80] Остальные вариации связаны с факторами окружающей среды и другими генами, влияющими на механизм заболевания.[19]
Ожидаемая продолжительность жизни при HD обычно составляет около 20 лет после появления видимых симптомов.[19] Большинство опасных для жизни осложнений возникает из-за координации мышц и, в меньшей степени, поведенческих изменений, вызванных снижением когнитивных функций. Самый большой риск пневмония, который вызывает смерть у одной трети людей с HD. Поскольку способность синхронизировать движения ухудшается, возникают трудности с очисткой легких и повышается риск всасывающий еда или питье увеличивают риск заражения пневмонией. Второй по величине риск - это сердечное заболевание, что является причиной почти четверти смертельных случаев среди больных HD.[19] Самоубийство является третьей по значимости причиной смертельных исходов: 7,3% людей с HD покончили с собой и до 27% пытались это сделать. Неясно, в какой степени на суицидальные мысли влияют поведенческие симптомы, поскольку они указывают на желание больного избежать более поздних стадий заболевания.[81][82][83] Другие связанные риски включают удушье, физическая травма от падений и недоедания.[19]
Эпидемиология
Позднее начало болезни Гентингтона означает, что она обычно не влияет на репродуктивную функцию.[19] Во всем мире распространенность HD составляет 5–10 случаев на 100 000 человек,[84][85] но сильно различается географически в результате этнической принадлежности, местной миграции и прошлых моделей иммиграции.[19] Распространенность одинакова для мужчин и женщин. Частота встречаемости наиболее высока в народы западноевропейского происхождения, в среднем около 7 на 100 000 человек, что ниже в остальном мире; например, один на миллион человек азиатского и африканского происхождения. Эпидемиологическое исследование распространенности болезни Хантингтона в Великобритании в период с 1990 по 2010 год в 2013 г. показало, что средняя распространенность в Великобритании составляла 12,3 на 100 000 человек.[19][86] Кроме того, в некоторых локализованных районах распространенность намного выше, чем в среднем по региону.[19] Один из самых высоких показателей заболеваемости - в изолированных популяциях Озеро Маракайбо регион Венесуэла, где HD затрагивает до 700 на 100 000 человек.[19][87] Другие области высокой локализации были обнаружены в Тасмания и конкретные регионы Шотландия, Уэльс и Швеция.[83] Повышенная распространенность в некоторых случаях происходит из-за местного эффект основателя, историческая миграция перевозчиков в область географическая изоляция.[83][88] Некоторые из этих носителей были прослежены сотни лет назад с использованием генеалогический исследования.[83] Генетический гаплотипы может также дать ключ к разгадке географических вариаций распространенности.[83][89] Исландия напротив, имеет довольно низкую распространенность - 1 на 100000, несмотря на то, что Исландцы как люди происходят от ранних германских племен Скандинавии, которые также дали начало Шведы; все случаи, за исключением одного, произошедшего почти два столетия назад, произошли от потомков пары, жившей в начале XIX века.[90] Финляндия также имеет низкий уровень заболеваемости - всего 2,2 на 100 000 человек.[91]
До открытия генетический тест, статистика может включать только клинический диагноз на основе физических симптомов и история семьи HD, за исключением тех, кто умер от других причин до постановки диагноза. Теперь эти случаи можно включать в статистику; и по мере того, как тест становится все более доступным, оценки распространенности и заболеваемости могут увеличиваться.[83][92]
История

Хотя болезнь Хантингтона была признана заболеванием по крайней мере с Средний возраст, причина была неизвестна до недавнего времени. Болезнь Хантингтона на протяжении всей своей истории носила разные имена, так как понимание болезни изменилось. Первоначально БГ называли просто «хореей» для отрывистых танцевальных движений, связанных с болезнью, но также называли «наследственной хореей» и «хронической прогрессирующей хореей».[94] Первое определенное упоминание HD было в письме Чарльз Оскар Уотерс, опубликовано в первом издании Робли Данглисон с Практика медицины в 1842 г. Уотерс описал «форму хореи, вульгарно называемую магрумом», включая точные описания хореи, ее прогрессирования и сильной наследственности.[95] В 1846 г. Чарльз Горман наблюдали, как более высокая распространенность, по-видимому, наблюдается в локализованных регионах.[95] Независимо от Гормана и Уотерса, оба ученика Данглисона в Медицинский колледж Джефферсона в Филадельфии,[96] Йохан Кристиан Лунд также произвел раннее описание в 1860 году.[95] Он особо отметил, что в Сетесдален, уединенная горная долина в Норвегия, была высокая распространенность деменции, связанной с паттерном подергивания двигательных расстройств, которые передавались в семьях.[97]
Первое подробное описание болезни было сделано Джордж Хантингтон в 1872 году. Изучая совокупный анамнез нескольких поколений семьи, демонстрирующей сходные симптомы, он понял, что их состояния должны быть связаны; он представил свое подробное и точное определение болезни в качестве своей первой статьи. Хантингтон описал точную схему наследования аутосомно-доминантного заболевания за много лет до повторного открытия учеными Менделирующее наследование.
О его наследственной природе. Когда один или оба родителя проявляют проявления болезни ... одно или несколько потомков почти неизменно страдают от болезни ... Но если по какой-то причине эти дети проживут жизнь без нее, нить оборвется, и внуки и правнуки первых шейкеров могут быть уверены, что они свободны от болезни.[93][98]
сэр Уильям Ослер интересовался расстройством и хореей в целом и был впечатлен статьей Хантингтона, в которой говорилось, что «в истории медицины есть несколько случаев, когда болезнь была описана более точно, более графически или более кратко».[95][99] Неизменный интерес Ослера к HD в сочетании с его влиянием в области медицины помогли быстро распространить осведомленность и знания о расстройстве среди медицинского сообщества.[95] Большой интерес проявили ученые Европы, в том числе Луи Теофиль Жозеф Ландузи, Дезире-Маглуар Бурневиль, Камилло Гольджи, и Джозеф Жюль Дежерин, и до конца века большая часть исследований HD была европейского происхождения.[95] К концу XIX века исследования и отчеты о HD были опубликованы во многих странах, и это заболевание было признано во всем мире.[95]
Во время повторного открытия менделевского наследования на рубеже 20-го века, HD был предварительно использован как пример аутосомно-доминантного наследования.[95] Английский биолог Уильям Бейтсон использовали родословные пострадавших семей, чтобы установить, что HD имеет аутосомно-доминантный образец наследования.[96] Сильный образец наследования побудил нескольких исследователей, в том числе Смит Эли Джеллифф, чтобы попытаться отследить и связать членов семьи предыдущих исследований.[95] Джеллифф собирал информацию со всех концов Нью-Йорк и опубликовал несколько статей о генеалогии HD в Новая Англия.[100] Исследование Джеллиффа вызвало интерес его друга по колледжу, Чарльз Давенпорт, который поручил Элизабет Манси провести первое полевое исследование Восточное побережье США семей с HD и построить их родословные.[101] Давенпорт использовал эту информацию, чтобы задокументировать переменный возраст начала и диапазон симптомов HD; он утверждал, что большинство случаев HD в США можно проследить до нескольких человек.[101] Это исследование было дополнено в 1932 г. П. Р. Весси, который популяризировал идею о том, что трое ушедших братьев Англия в 1630 г. Бостон были прародителями HD в США.[102] Утверждение, что самые ранние предки были установлены и евгенический предвзятость работы Манси, Давенпорта и Весси способствовала недопониманию и предубеждениям в отношении HD.[96] Манси и Давенпорт также популяризировали идею о том, что в прошлом некоторые люди, страдающие HD, могли считаться одержимыми духами или жертвами колдовство, а иногда были избегали или же сослан обществом.[103][104] Эта идея не получила подтверждения. Исследователи нашли противоположные доказательства; например, сообщество семьи, изученное Джорджем Хантингтоном, открыто принимало тех, кто проявлял симптомы HD.[96][103]
Поиск причины этого состояния был значительно усилен в 1968 году, когда Фонд наследственных болезней (HDF) был создан Милтон Векслер, а психоаналитик основанный в Лос-Анджелес, Калифорния, чьей жене Леоноре Сабин ранее в том же году был поставлен диагноз болезнь Хантингтона.[105] Трое братьев жены Векслера также страдали этим заболеванием.
Фонд участвовал в привлечении более 100 ученых в Американо-венесуэльский совместный проект по болезни Хантингтона который в течение 10-летнего периода с 1979 года работал над локализацией генетической причины.[106] Это было достигнуто в 1983 году, когда был приблизительно обнаружен причинный ген,[88] а в 1993 году ген был точно расположен на хромосоме 4 (4p16.3).[107] Исследование было сосредоточено на популяциях двух изолированных Венесуэльский деревни Барранкитас и Лагунетас, где была необычно высокая распространенность болезни. В нем приняли участие более 18 000 человек, в основном из одной большой семьи, и в результате HD стал первым аутосомный болезнь локус найдено с помощью анализ генетической связи.[107][108] Среди прочих нововведений проект разработан ДНК-маркировка методы, которые были важным шагом на пути к Проект "Геном человека" возможный.[106]
В то же время были сделаны ключевые открытия, касающиеся механизмов расстройства, в том числе открытия Анита Хардинг группа исследователей влияния длины гена.[109]
Моделирование болезни у различных видов животных, таких как трансгенный мышь, разработанная в 1996 году, позволила проводить более масштабные эксперименты. Как у этих животных быстрее метаболизм и продолжительность жизни намного короче, чем у людей, результаты экспериментов получены раньше, что ускоряет исследования. Открытие 1997 года, что фрагменты mhtt неправильно сложить привели к открытию ядерных включений, которые они вызывают. Эти достижения привели к все более обширным исследованиям белков, связанных с заболеванием, потенциальных лекарств, методов ухода и самого гена.[95][110]
Ранее это состояние называлось «хореей Хантингтона», но этот термин был заменен на «болезнь Хантингтона», потому что не у всех пациентов развивается хорея и из-за важности когнитивных и поведенческих проблем.[111]
Общество и культура
Этика
Болезнь Хантингтона, особенно применение генетического теста на болезнь, подняла несколько этических проблем. Вопросы генетического тестирования включают определение того, насколько зрелым должен быть человек, прежде чем он будет признан подходящим для тестирования, обеспечение конфиденциальности результатов и следует ли разрешить компаниям использовать результаты тестов для принятия решений о приеме на работу, страховании жизни или других финансовых вопросах. Были споры, когда Чарльз Давенпорт предложил в 1910 г. принудительная стерилизация и иммиграция контроль будет использоваться для людей с определенными заболеваниями, включая HD, как часть евгеника движение.[112] Экстракорпоральное оплодотворение имеет некоторые проблемы с использованием эмбрионов. Некоторые исследования HD имеют этические проблемы из-за использования тестирование животных и эмбриональные стволовые клетки.[113][114]
Разработка точного диагностического теста на болезнь Хантингтона вызвала социальные, юридические и этические проблемы, связанные с доступом к результатам исследования человека и их использованием.[115][116]Во многих руководствах и процедурах тестирования предусмотрены строгие процедуры раскрытия информации и конфиденциальности, позволяющие людям решать, когда и как получить свои результаты, а также кому они предоставляются.[19] Финансовые учреждения и предприятия сталкиваются с вопросом, использовать ли результаты генетических тестов при оценке человека, например, для страхования жизни или трудоустройства. Страховые компании Соединенного Королевства согласились с Департамент здравоохранения и социальной защиты что до 2017 года клиентам не нужно будет раскрывать им прогностические генетические тесты, но это соглашение явно исключает одобренный правительством тест для Хантингтона при написании полисов со значением более 500 000 фунтов стерлингов.[117][118] Как и в случае с другими неизлечимыми генетическими состояниями с более поздним началом, этически сомнительно проводить предсимптоматическое тестирование ребенка или подростка, поскольку для этого человека не будет никакой медицинской пользы. Существует консенсус в отношении тестирования только тех людей, которые считаются когнитивно зрелыми, хотя есть контраргумент, что родители имеют право принимать решение от имени своего ребенка. При отсутствии эффективного лечения тестирование человека совершеннолетие кто не считается компетентный в большинстве случаев считается неэтичным.[39][119][120]
Есть этические проблемы, связанные с пренатальное генетическое тестирование или же предимплантационная генетическая диагностика чтобы гарантировать, что ребенок не родился с данным заболеванием.[121] Например, пренатальное тестирование поднимает вопрос о выборочном аборте, который некоторые считают неприемлемым.[121] Поскольку это преобладающее заболевание, возникают трудности в ситуациях, когда родитель не хочет знать свой собственный диагноз. Это потребует, чтобы некоторые части процесса были в секрете от родителя.[121]
Поддерживающие организации

В 1968 году, после перенесенного HD в семье своей жены, доктор Милтон Векслер был вдохновлен начать Фонд наследственных болезней (HDF) с целью лечения генетических заболеваний путем координации и поддержки исследований.[11] Фонд и дочь Векслера, Нэнси Векслер, были ключевыми частями исследовательской группы в Венесуэле, открывшей ген HD.[11]
Примерно в то же время, когда образовалась HDF, Марджори Гатри помог основать Комитет по борьбе с болезнью Хантингтона (ныне Общество болезни Хантингтона Америки ), после мужа Вуди Гатри умер от осложнений HD.[12]
С тех пор во многих странах мира сформировались исследовательские организации и организации, которые помогли повысить осведомленность общественности о HD. Некоторые из них сотрудничают в зонтичных организациях, таких как Международная ассоциация Хантингтона и Европейская сеть HD.[122] Многие организации поддержки проводят ежегодные информационные мероприятия по HD, некоторые из которых были одобрены их правительствами. Например, 6 июня объявлено Национальным днём распространения информации о болезни Хантингтона. Сенат США.[123]
Крупнейший спонсор исследований болезни Хантингтона в мире,[124] это Фонд Инициативы по борьбе с болезнью Хантингтона (CHDI), США некоммерческий биомедицинский фонд, целью которого является «быстрое открытие и разработка лекарств, замедляющих или замедляющих болезнь Хантингтона».[125] CHDI ранее назывался High Q Foundation. В 2006 году он потратил 50 миллионов долларов на исследования болезни Хантингтона.[124] CHDI сотрудничает со многими академическими и коммерческими лабораториями по всему миру и занимается надзором и управлением исследовательскими проектами, а также финансированием.[126] Многие организации существуют для поддержки и информирования тех, кто пострадал от HD, в том числе Ассоциация болезни Хантингтона в Соединенном Королевстве.
Направления исследований
Исследования механизма HD сосредоточены на выявлении функционирования HTT, того, как mhtt отличается или мешает ему, а также патологии головного мозга, которую вызывает это заболевание.[127] Исследования проводятся с использованием in vitro методы, модели на животных и добровольцы. Модели на животных имеют решающее значение для понимания фундаментальных механизмов, вызывающих болезнь, и для поддержки ранних стадий заболевания. разработка лекарств.[110] Животные с химически индуцированным повреждением головного мозга проявляют HD-подобные симптомы и первоначально использовались, но они не имитировали прогрессирующие признаки заболевания.[128] Идентификация причинного гена позволила разработать многие трансгенное животное модели в том числе нематода черви, Дрозофила плодовые мошки, мыши, крысы, овцы, свиньи и обезьяны, которые экспрессируют мутантный хантингтин и развивают прогрессивный нейродегенерация и HD-подобные симптомы.[110]
В настоящее время проводятся исследования множества различных подходов к предотвращению болезни Хантингтона или замедлению ее прогрессирования.[127] Стратегии модификации заболевания можно в общих чертах сгруппировать в три категории: снижение уровня мутантного белка хантингтина (включая сплайсинг генов и подавление гена ); подходы, направленные на повышение выживаемости нейронов за счет снижения вреда, наносимого белком конкретным клеточным путям и механизмам (включая белковый гомеостаз и гистоновая деацетилаза торможение); и стратегии по замене потерянных нейронов. Кроме того, в стадии разработки находятся новые методы лечения, улучшающие работу мозга; они направлены на создание симптоматической, а не изменяющей болезнь терапии, и включают ингибиторы фосфодиэстеразы.[129][130]
В 2020 году CHDI Foundation начал сотрудничество в области вычислительных исследований малых молекул с OpenEye Scientific фокусируясь на лечении с помощью малых молекул, используя платформу молекулярного дизайна OpenEye, известную как Орион.[125]
Снижение производства хантинтина
Подавление гена направлена на снижение продукции мутантного белка, поскольку HD вызывается единственный доминантный ген кодирует токсичный белок. Эксперименты по подавлению гена на моделях мышей показали, что когда экспрессия mhtt снижается, симптомы улучшаются.[131] Безопасность РНК-интерференция, и аллель-специфический олигонуклеотид (ASO) методы подавления гена были продемонстрированы на мышах и в головном мозге крупных приматов макака.[132][133] Аллел-специфическое молчание пытается заставить замолчать мутантный htt, оставляя HTT дикого типа нетронутым. Одним из способов достижения этого является идентификация полиморфизмов, присутствующих только на одном аллеле, и получение препаратов для подавления гена, нацеленных на полиморфизмы только в мутантном аллеле.[134] Первое испытание по сайленсингу генов с участием людей с HD началось в 2015 году, и в нем проверялась безопасность IONIS-HTTRx, произведенного Ionis Pharmaceuticals и во главе с UCL Институт неврологии.[135][136] Мутантный хантингтин был впервые обнаружен и количественно определен в спинномозговая жидкость от носителей мутации Хантингтона в 2015 году с использованием нового метода «подсчета одиночных молекул» иммуноанализ,[137] обеспечение прямого способа оценки достижения желаемого эффекта при лечении хантинтин-снижающих препаратов.[138][139] По аналогии, сплайсинг генов изучаются методы, чтобы попытаться восстановить геном с ошибочным геном, вызывающим HD, с использованием таких инструментов, как CRISPR / Cas9.[130]
Увеличение клиренса хантинтина
Еще одна стратегия снижения уровней мутантного хантингтина - это увеличение скорости, с которой клетки способны очищать мутантный белок.[140] Поскольку мутантный белок хантингтин (и многие другие склонные к агрегированию белки) разрушается в результате аутофагии, повышение уровня аутофагии может снизить уровни токсичного белка и тем самым облегчить течение болезни.[141] Фармакологические и генетические индукторы аутофагии были протестированы на различных моделях болезни Хантингтона, и было показано, что многие из них снижают уровни mHTT и уменьшают токсичность.[140]
Повышение выживаемости клеток
Среди подходов, направленных на повышение выживаемости клеток в присутствии мутантного хантингтина, можно выделить коррекцию транскрипционная регуляция с помощью ингибиторы гистондеацетилазы, модулирующий агрегирование хантинтина, улучшающего метаболизм и митохондриальная функция и восстанавливая функцию синапсы.[131]
Замена нейронов
Стволовыми клетками замена поврежденных нейронов трансплантацией стволовые клетки в пораженные участки мозга. Эксперименты дали смешанные результаты с использованием этого метода на животных моделях и предварительных исследованиях человека. клинические испытания.[142] Каким бы ни был их терапевтический потенциал в будущем, стволовые клетки уже являются ценным инструментом для изучения болезни Хантингтона в лаборатории.[143]
Клинические испытания
В 2020 году их было 197 клинические испытания относящиеся к различным методам лечения и биомаркерам болезни Хантингтона, перечисленным как текущие, набираемые или недавно завершенные.[144]
Соединения испытанный, которые не смогли предотвратить или замедлить прогрессирование болезни Хантингтона, включают Ремасемид, Коэнзим Q10, рилузол, креатин, миноциклин, этил-EPA, фенилбутират и димебон.[145]
Смотрите также
 Портал медицины
Портал медицины
Рекомендации
- ^ а б c d е ж грамм час я j k л Даялу П., Альбин Р.Л. (февраль 2015 г.). «Болезнь Хантингтона: патогенез и лечение». Неврологические клиники. 33 (1): 101–14. Дои:10.1016 / j.ncl.2014.09.003. PMID 25432725.
- ^ а б c d е ж грамм Кэрон Н.С., Райт Г.Е., Хайден М.Р. (2020). Адам М.П., Ардингер Х.Х., Пагон Р.А., Уоллес С.Е., Бин Л.Дж., Стивенс К., Амемия А. (ред.). «Болезнь Хантингтона». GeneReviews. PMID 20301482.
- ^ а б c d е ж грамм час я j k л Фрэнк С. (январь 2014 г.). «Лечение болезни Гентингтона». Нейротерапия. 11 (1): 153–60. Дои:10.1007 / s13311-013-0244-z. ЧВК 3899480. PMID 24366610.
- ^ а б c d е ж грамм час "Информационная страница о болезни Хантингтона | Национальный институт неврологических заболеваний и инсульта". www.ninds.nih.gov. Получено 14 декабря 2020.
- ^ а б Дурр А., Гарджуло М., Фейнгольд Дж. (Ноябрь 2012 г.). «Пресимптоматическая фаза болезни Хантингтона». Revue Neurologique. 168 (11): 806–8. Дои:10.1016 / j.neurol.2012.07.003. PMID 22902173.
- ^ Ферри, Фред Ф. (2010). Дифференциальный диагноз Ферри: практическое руководство по дифференциальной диагностике симптомов, признаков и клинических расстройств (2-е изд.). Филадельфия, Пенсильвания: Эльзевьер / Мосби. п. Глава H. ISBN 978-0323076999.
- ^ а б «Молекулярный патогенез при болезни Хантингтона». Protein.bio.msu.ru. Получено 8 ноября 2020.
- ^ а б c d е Бейтс ГП, Дорси Р., Гуселла Дж. Ф., Хайден М. Р., Кей С., Ливитт Б. Р., Нэнси М., Росс К. А., Скахилл Р. И., Ветцель Р., Уайлд Э. Дж., Тебризи SJ (Апрель 2015 г.). «Болезнь Хантингтона». Nature Reviews Праймеры от болезней. 1: 15005. Дои:10.1038 / nrdp.2015.5. PMID 27188817. S2CID 25759303.
- ^ а б Вале ТК, Кардосо Ф (2015). «Хорея: Путешествие по истории». Тремор и другие гиперкинетические движения. 5. Дои:10.7916 / D8WM1C98. ЧВК 4454991. PMID 26056609.
- ^ а б "Изучение болезни Хантингтона". www.genome.gov. В архиве из оригинала 4 июля 2016 г.. Получено 19 июля 2016.
- ^ а б c d «История ХДФ». Фонд наследственных болезней. Архивировано из оригинал 19 ноября 2015 г.. Получено 18 ноября 2015.
- ^ а б "История и генетика болезни Хантингтона | Американское общество болезни Хантингтона". Получено 14 декабря 2020.
- ^ а б c d ван Дуйн Э., Кингма Э.М., ван дер Маст RC (2007). «Психопатология у проверенных носителей гена болезни Гентингтона». Журнал нейропсихиатрии и клинической неврологии. 19 (4): 441–8. Дои:10.1176 / appi.neuropsych.19.4.441. PMID 18070848.
- ^ "Болезнь Хантингтона". www.nhsinform.scot. Получено 12 июля 2020.
- ^ а б c «Болезнь Хантингтона». Genreviews книжная полка. Вашингтонский университет. Июнь 2020 г.. Получено 22 ноября 2020.
- ^ Диагностическое и статистическое руководство психических расстройств: DSM-5 (5-е изд.). Арлингтон, Вирджиния: Американская психиатрическая ассоциация. 2013. с. 639. ISBN 9780890425541.
- ^ а б Кремер Б (2002). «Клиническая неврология болезни Хантингтона». В Bates G, Harper P, Jones L (ред.). Болезнь Хантингтона - Третье издание. Оксфорд: Издательство Оксфордского университета. С. 28–53. ISBN 978-0-19-851060-4.
- ^ Wagle AC, Wagle SA, Marková IS, Berrios GE (2000). «Психиатрическая заболеваемость при болезни Хантингтона». Неврология, психиатрия и исследования мозга (8): 5–16.
- ^ а б c d е ж грамм час я j k л м п о п q р s т ты v ш Икс у z аа ab ac объявление ае аф аг ах ай эй ак аль являюсь ан ао ap водный ар в качестве в au средний ау топор ай az ба bb до н.э bd быть парень bg бх би Ъ bk бл бм млрд бо бп Уокер Ф.О. (январь 2007 г.). "Болезнь Хантингтона". Ланцет. 369 (9557): 218–28. Дои:10.1016 / S0140-6736 (07) 60111-1. PMID 17240289. S2CID 46151626.
- ^ а б c d е ж грамм Montoya A, Price BH, Menear M, Lepage M (январь 2006 г.). «Визуализация мозга и когнитивные дисфункции при болезни Хантингтона» (PDF). Журнал психиатрии и неврологии. 31 (1): 21–9. ЧВК 1325063. PMID 16496032. Архивировано из оригинал (PDF) 23 марта 2016 г.. Получено 17 сентября 2008.
- ^ а б Дики А.С., La Spada AR (апрель 2018 г.). «Развитие терапии при болезни Хантингтона: от текущих стратегий к новым возможностям». Американский журнал медицинской генетики. Часть А. 176 (4): 842–861. Дои:10.1002 / ajmg.a.38494. ЧВК 5975251. PMID 29218782.
- ^ Азиз Н.А., ван дер Марк М.А., Пейл Х., Олде Риккерт М.Г., Блум Б.Р., Роос Р.А. (декабрь 2008 г.). «Похудание при нейродегенеративных расстройствах». Журнал неврологии. 255 (12): 1872–80. Дои:10.1007 / s00415-009-0062-8. PMID 19165531. S2CID 26109381.
- ^ "Буклет Хантингтонского общества Канады" (PDF). Справочник по уходу за продвинутой стадией болезни Хантингтона. HD Общество Канады. 11 апреля 2007 г. Архивировано с оригинал (PDF) 25 июня 2008 г.. Получено 10 августа 2008.
- ^ Мюррей ЭД, Баттнер Н., Прайс Б.Н. (2012). «Депрессия и психоз в неврологической практике». В Bradley WG, Daroff RB, Fenichel GM, Jankovic J (ред.). Неврология Брэдли в клинической практике (6-е изд.). Филадельфия, Пенсильвания: Эльзевьер / Сондерс. п. 108. ISBN 978-1-4377-0434-1.
- ^ ван дер Бург Дж. М., Бьёркквист М., Брундин П. (август 2009 г.). «За пределами мозга: широко распространенная патология при болезни Хантингтона». Ланцет. Неврология. 8 (8): 765–74. Дои:10.1016 / S1474-4422 (09) 70178-4. PMID 19608102. S2CID 14419437.
- ^ Кацуно М, Банно Х, Судзуки К., Такеучи Й, Кавасима М, Танака Ф, Адачи Х, Собуэ Дж. (Май 2008 г.). «Молекулярная генетика и биомаркеры полиглутаминовых заболеваний». Современная молекулярная медицина. 8 (3): 221–34. Дои:10.2174/156652408784221298. PMID 18473821.
- ^ Сквитери Ф., Фрати Л., Чиармиелло А., Ластория С., Куоррелл О. (февраль 2006 г.). «Ювенильная болезнь Хантингтона: отличается ли патогенетический механизм дозирования от классической взрослой болезни?». Механизмы старения и развития. 127 (2): 208–12. Дои:10.1016 / j.mad.2005.09.012. PMID 16274727. S2CID 20523093.
- ^ Нэнси М.А., Майерс Р.Х. (2001). «Болезнь Хантингтона с юношеским началом - клинические и исследовательские перспективы». Обзоры исследований в области умственной отсталости и пороков развития. 7 (3): 153–7. Дои:10.1002 / mrdd.1022. PMID 11553930.
- ^ Пассарж Э (2001). Цветовой атлас генетики (2-е изд.). Тиме. п.142. ISBN 978-0-86577-958-7.
- ^ Ридли Р.М., Фрит С.Д., Ворон Т.Дж., Коннелли П.М. (сентябрь 1988 г.). «Ожидание болезни Гентингтона передается по мужской линии, но может происходить от женщины». Журнал медицинской генетики. 25 (9): 589–95. Дои:10.1136 / jmg.25.9.589. ЧВК 1051535. PMID 2972838.
- ^ Семака А., Крейтон С., Варби С., Хайден М. Р. (октябрь 2006 г.). «Прогностическое тестирование болезни Хантингтона: интерпретация и значение промежуточных аллелей». Клиническая генетика. 70 (4): 283–94. Дои:10.1111 / j.1399-0004.2006.00668.x. PMID 16965319. S2CID 26007984.
- ^ Wexler NS, Young AB, Tanzi RE, Travers H, Starosta-Rubinstein S, Penney JB, Snodgrass SR, Shoulson I, Gomez F, Ramos Arroyo MA (1987). «Гомозиготы по болезни Хантингтона» (PDF). Природа. 326 (6109): 194–7. Bibcode:1987 Натур. 326..194 Вт. Дои:10.1038 / 326194a0. HDL:2027.42/62543. PMID 2881213. S2CID 4312171.
- ^ Сквитери Ф., Геллера С., Каннелла М., Мариотти С., Цислаги Дж., Рубинштейн Д.К., Альмквист Е.В., Тернер Д., Бахуд-Леви А.С., Симпсон С.А., Делатики М., Маглионе В., Хайден М.Р., Донато С.Д. (апрель 2003 г.) «Гомозиготность по мутации CAG при болезни Хантингтона связана с более тяжелым клиническим течением». Мозг. 126 (Pt 4): 946–55. Дои:10.1093 / мозг / awg077. PMID 12615650.
- ^ Goehler H, Lalowski M, Stelzl U, Waelter S, Stroedicke M, Worm U, Droege A, Lindenberg KS, Knoblich M, Haenig C, Herbst M, Suopanki J, Scherzinger E, Abraham C, Bauer B, Hasenbank R, Fritzsche , Ludewig AH, Büssow K, Buessow K, Coleman SH, Gutekunst CA, Landwehrmeyer BG, Lehrach H, Wanker EE (сентябрь 2004 г.). «Сеть взаимодействия белков связывает GIT1, усилитель агрегации хантингтина, с болезнью Хантингтона». Молекулярная клетка. 15 (6): 853–65. Дои:10.1016 / j.molcel.2004.09.016. PMID 15383276.
- ^ Глайч К.Е., Садри-Вакили Г. (2015). «Эпигенетические механизмы, участвующие в патогенезе болезни Хантингтона». Журнал болезни Хантингтона. 4 (1): 1–15. Дои:10.3233 / JHD-159001. PMID 25813218.
- ^ Харджес П., Ванкер Э. Э. (август 2003 г.). «Функция охоты на хантингтин: партнеры по взаимодействию рассказывают много разных историй». Тенденции в биохимических науках. 28 (8): 425–33. Дои:10.1016 / S0968-0004 (03) 00168-3. PMID 12932731.
- ^ а б c Cattaneo E, Zuccato C, Tartari M (декабрь 2005 г.). «Нормальная функция хантингтина: альтернативный подход к болезни Хантингтона». Обзоры природы Неврология. 6 (12): 919–30. Дои:10.1038 / nrn1806. PMID 16288298. S2CID 10119487.
- ^ а б c d е Рубинштейн, округ Колумбия, Кармайкл Дж (август 2003 г.). «Болезнь Хантингтона: молекулярные основы нейродегенерации». Обзоры экспертов в области молекулярной медицины. 5 (20): 1–21. Дои:10.1017 / S1462399403006549. PMID 14585171.
- ^ а б Блох М., Хайден М.Р. (январь 1990 г.). «Мнение: прогностическое тестирование на болезнь Хантингтона в детстве: проблемы и последствия». Американский журнал генетики человека. 46 (1): 1–4. ЧВК 1683548. PMID 2136787.
- ^ а б c Садри-Вакили Дж., Ча Дж. Х. (июнь 2006 г.). «Механизмы болезни: модификации гистонов при болезни Хантингтона». Природа Клиническая Практика Неврология. 2 (6): 330–8. Дои:10.1038 / ncpneuro0199. PMID 16932577. S2CID 12474262.
- ^ Лю З., Чжоу Т., Зиглер А.С., Димитрион П., Цзо Л. (2017). «Окислительный стресс при нейродегенеративных заболеваниях: от молекулярных механизмов до клинического применения». Окислительная медицина и клеточное долголетие. 2017: 2525967. Дои:10.1155/2017/2525967. ЧВК 5529664. PMID 28785371.
- ^ Кумар А., Ратан Р. Р. (октябрь 2016 г.). «Окислительный стресс и болезнь Хантингтона: хорошее, плохое и уродливое». Журнал болезни Хантингтона. 5 (3): 217–237. Дои:10.3233 / JHD-160205. ЧВК 5310831. PMID 27662334.
- ^ Первес Д., Огюстин Г. А., Фицпатрик Д., Холл В., Ламантия А. С., Макнамара Д. О., Уильямс С. М. (2001). «Модуляция движения базальных ганглиев - контуров в системе базальных ганглиев». В Purves D (ред.). Неврология (2-е изд.). Сандерленд, Массачусетс: Sinauer Associates. ISBN 978-0-87893-742-4. В архиве из оригинала 18 февраля 2009 г.. Получено 1 апреля 2009.
- ^ Lobsiger CS, Cleveland DW (ноябрь 2007 г.). «Глиальные клетки как внутренние компоненты неавтономного нейродегенеративного заболевания». Природа Неврология. 10 (11): 1355–60. Дои:10.1038 / nn1988. ЧВК 3110080. PMID 17965655.
- ^ а б Кроссман AR (май 2000 г.). «Функциональная анатомия двигательных нарушений». Журнал анатомии. 196 (Pt 4) (4): 519–25. Дои:10.1046 / j.1469-7580.2000.19640519.x. ЧВК 1468094. PMID 10923984.
- ^ Даффи Дж (2013). Нарушения моторной речи: субстраты, дифференциальная диагностика и лечение (3-е изд.). Сент-Луис, Миссури: Эльзевир. С. 196–7.
- ^ а б Петруска Дж, Хартенстайн М.Дж., Гудман М.Ф. (февраль 1998 г.). «Анализ проскальзывания цепи при расширении ДНК-полимеразы триплетных повторов CAG / CTG, связанных с нейродегенеративным заболеванием». Журнал биологической химии. 273 (9): 5204–10. Дои:10.1074 / jbc.273.9.5204. PMID 9478975.
- ^ Стеффан Дж. С., Бодаи Л., Паллос Дж., Полман М., МакКэмпбелл А., Апостол Б. Л. и др. (Октябрь 2001 г.). «Ингибиторы гистон-деацетилазы останавливают полиглутамин-зависимую нейродегенерацию у дрозофилы». Природа. 413 (6857): 739–43. Bibcode:2001Натура.413..739S. Дои:10.1038/35099568. PMID 11607033. S2CID 4419980.
- ^ Гайяр Ф (1 мая 2007 г.). "Болезнь Хантингтона". Радиологическая картина дня. www.radpod.org. Архивировано из оригинал 22 октября 2007 г.. Получено 24 июля 2009.
- ^ Рао А.К., Муратори Л., Луи Э.Д., Московиц С.Б., Мардер К.С. (апрель 2009 г.). «Клиническое измерение нарушений подвижности и равновесия при болезни Хантингтона: достоверность и отзывчивость». Походка и поза. 29 (3): 433–6. Дои:10.1016 / j.gaitpost.2008.11.002. PMID 19111470.
- ^ «Единая шкала оценки болезни Хантингтона (UHDRS)». UHDRS и база данных. HSG. 1 февраля 2009 г. В архиве с оригинала 11 августа 2015 г.. Получено 14 апреля 2009.
- ^ Майерс Р.Х. (апрель 2004 г.). «Генетика болезни Хантингтона». NeuroRx. 1 (2): 255–62. Дои:10.1602 / Neurorx.1.2.255. ЧВК 534940. PMID 15717026.
- ^ а б c d е ж грамм de Die-Smulders CE, de Wert GM, Liebaers I, Tibben A, Evers-Kiebooms G (май 2013 г.). «Репродуктивные возможности для будущих родителей в семьях с болезнью Гентингтона: клинические, психологические и этические размышления». Обновление репродукции человека. 19 (3): 304–15. Дои:10.1093 / humupd / dms058. PMID 23377865. de Die-Smulders CE, de Wert GM, Liebaers I, Tibben A, Evers-Kiebooms G (2013). «Репродуктивные возможности для будущих родителей в семьях с болезнью Гентингтона: клинические, психологические и этические размышления». Обновление репродукции человека. 19 (3): 304–15. Дои:10.1093 / humupd / dms058. PMID 23377865.
- ^ Форрест Кинан К., Симпсон С.А., Мендзыбродзка З., Александр Д.А., Семпер Дж. (Июнь 2013 г.). «Как партнеры узнают о риске болезни Хантингтона в супружеских отношениях?». Журнал генетического консультирования. 22 (3): 336–44. Дои:10.1007 / s10897-012-9562-2. PMID 23297124. S2CID 15447709.
- ^ Эрвин С., Уильямс Дж. К., Джул А. Р., Менгелинг М., Миллс Дж. А., Бомбард И., Хайден М. Р., Куэйд К., Шоулсон И., Тейлор С., Полсен Дж. С. (июль 2010 г.). «Восприятие, опыт и реакция на генетическую дискриминацию при болезни Хантингтона: международное исследование RESPOND-HD». Американский журнал медицинской генетики. Часть B, Психоневрологическая генетика. 153B (5): 1081–93. Дои:10.1002 / ajmg.b.31079. ЧВК 3593716. PMID 20468061.
- ^ Бурсон CM, Марки KR (сентябрь 2001 г.). «Вопросы генетического консультирования в прогнозирующем генетическом тестировании семейных неврологических заболеваний у взрослых». Семинары по детской неврологии. 8 (3): 177–86. Дои:10.1053 / spen.2001.26451. PMID 11575847.
- ^ Смит Дж. А., Мичи С., Стивенсон М., Куоррелл О. (март 2002 г.). "Восприятие риска и процессы принятия решений у кандидатов на генетическое тестирование на болезнь Хантингтона: интерпретативный феноменологический анализ". Журнал психологии здоровья. 7 (2): 131–44. Дои:10.1177/1359105302007002398. PMID 22114233. S2CID 40182214.
- ^ а б Хайден MR (март 2003 г.). «Прогностическое тестирование на болезнь Гентингтона: универсальная модель?». Ланцет. Неврология. 2 (3): 141–2. Дои:10.1016 / S1474-4422 (03) 00317-X. PMID 12849232. S2CID 39581496.
- ^ «Руководство по молекулярно-генетическому прогностическому тесту при болезни Гентингтона. Международная ассоциация Хантингтона (IHA) и Всемирная федерация неврологии (WFN), исследовательская группа по хореи Хантингтона». Неврология. 44 (8): 1533–6. Август 1994 г. Дои:10.1212 / WNL.44.8.1533. PMID 8058167.
- ^ Лосекут М., ван Бельзен М.Дж., Сенека С., Бауэр П., Стенхаус С.А., Бартон Д.Е. (май 2013 г.). «Рекомендации EMQN / CMGS по передовой практике молекулярно-генетического тестирования болезни Хантингтона». Европейский журнал генетики человека. 21 (5): 480–6. Дои:10.1038 / ejhg.2012.200. ЧВК 3641377. PMID 22990145.
- ^ Шульман JD, Black SH, Handyside A, Nance WE (февраль 1996 г.). «Преимплантационное генетическое тестирование на болезнь Хантингтона и некоторые другие доминантно наследуемые расстройства». Клиническая генетика. 49 (2): 57–8. Дои:10.1111 / j.1399-0004.1996.tb04327.x. PMID 8740912. S2CID 45703511.
- ^ Stern HJ, Harton GL, Sisson ME, Jones SL, Fallon LA, Thorsell LP, Getlinger ME, Black SH, Schulman JD (июнь 2002 г.). «Нераскрытие доимплантационной генетической диагностики болезни Хантингтона». Пренатальная диагностика. 22 (6): 503–7. Дои:10.1002 / pd.359. PMID 12116316. S2CID 33967835.
- ^ «Прогностическое тестирование на болезнь Хантингтона». 2011. В архиве из оригинала 22 января 2013 г.. Получено 7 мая 2013.
- ^ Кулиев А., Верлинский Ю. (апрель 2005 г.). «Преимплантационная диагностика: реальный вариант для вспомогательной репродукции и генетической практики». Текущее мнение в области акушерства и гинекологии. 17 (2): 179–83. Дои:10.1097 / 01.gco.0000162189.76349.c5. PMID 15758612. S2CID 9382420.
- ^ «Рекомендации по генетическому тестированию на болезнь Хантингтона». Фонд болезней наследственности. Архивировано из оригинал 26 июня 2015 г.. Получено 7 мая 2013.
- ^ а б Шнайдер С.А., Уокер Р.Х., Бхатия КП (сентябрь 2007 г.). «Синдромы, подобные болезни Хантингтона: что следует учитывать у пациентов с отрицательным результатом генного теста на болезнь Хантингтона». Природа Клиническая Практика Неврология. 3 (9): 517–25. Дои:10.1038 / ncpneuro0606. PMID 17805246. S2CID 9052603.
- ^ Франк С., Янкович Дж. (Март 2010 г.). «Достижения в фармакологическом лечении болезни Хантингтона». Наркотики. 70 (5): 561–71. Дои:10.2165/11534430-000000000-00000. PMID 20329804. S2CID 42386743. Архивировано из оригинал 8 октября 2011 г.
- ^ а б c Бонелли Р.М., Веннинг Г.К., Kapfhammer HP (март 2004 г.). «Болезнь Хантингтона: современные методы лечения и будущие терапевтические методы». Международная клиническая психофармакология. 19 (2): 51–62. Дои:10.1097/00004850-200403000-00001. PMID 15076012. S2CID 1956458.
- ^ Панайотакис PH, ДиСарио Дж.А., Хильден К., Огара М., Фанг Дж.С. (2008). «Установка трубки DPEJ предотвращает аспирационную пневмонию у пациентов из группы высокого риска». Питание в клинической практике. 23 (2): 172–5. Дои:10.1177/0884533608314537. PMID 18390785.
- ^ а б "Руководство по физиотерапии EHDN" (PDF). Европейская рабочая группа по физиотерапии сети HD. Архивировано из оригинал (PDF) 4 марта 2016 г.. Получено 15 ноября 2015.
- ^ Куин Л., Бузи М. (февраль 2012 г.). «Разработка руководств по физиотерапии и основанных на лечении классификаций для людей с болезнью Гентингтона». Ведение нейродегенеративных заболеваний. 2 (1): 21–31. Дои:10.2217 / нмт.11.86.
- ^ Халил Х., Куинн Л., ван Дерсен Р., Мартин Р., Россер А., Бусс М. (январь 2012 г.). «Приверженность использованию DVD с домашними упражнениями у людей с болезнью Гентингтона: точки зрения участников». Физиотерапия. 92 (1): 69–82. Дои:10.2522 / ptj.20100438. PMID 21960468.
- ^ Трэверс Э., Джонс К., Никол Дж. (2007). «Оказание паллиативной помощи при болезни Хантингтона». Международный журнал паллиативного ухода. 13 (3): 125–30. Дои:10.12968 / ijpn.2007.13.3.23274. PMID 17505405.
- ^ «FDA одобрило первый препарат для лечения хореи при болезни Хантингтона». Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США. 15 августа 2008 г. В архиве из оригинала 21 августа 2008 г.. Получено 10 августа 2008.
- ^ Морси С., Халил С.М., Дохейм М.Ф., Камель М.Г., Эль-Басиони Д.С., Ахмед Хассан Х.И. и др. (Август 2019 г.). «Эффективность этил-ЭПК в качестве лечения болезни Хантингтона: систематический обзор и метаанализ». Acta Neuropsychiatrica. 31 (4): 175–185. Дои:10.1017 / neu.2019.11. HDL:10069/39427. PMID 30890195. S2CID 84183892.
- ^ Исследования, Центр оценки лекарственных средств (17 июля 2019 г.). «В погоне за поздней дискинезией: революционное обозначение и одобрение вальбеназина». FDA. Получено 15 ноября 2020.
- ^ Цитром L (апрель 2016 г.). «Прорывные препараты на стыке психиатрии и неврологии». Международный журнал клинической практики. 70 (4): 298–9. Дои:10.1111 / ijcp.12805. PMID 27028671. S2CID 38537781.
- ^ Харпер П. (2002). «Генетическое консультирование и досимптомное тестирование». В Bates G, Harper P, Jones L (ред.). Болезнь Хантингтона - Третье издание. Оксфорд: Издательство Оксфордского университета. С. 198–242. ISBN 978-0-19-851060-4.
- ^ Харпер PS (июнь 1999 г.). «Болезнь Хантингтона: клиническая, генетическая и молекулярная модель нарушений с полиглутаминовыми повторами». Философские труды Лондонского королевского общества. Серия B, Биологические науки. 354 (1386): 957–61. Дои:10.1098 / rstb.1999.0446. ЧВК 1692597. PMID 10434293.
- ^ Эндрю С.Е., Голдберг Ю.П., Кремер Б., Телениус Х., Тейлманн Дж., Адам С., Старр Э., Сквитери Ф., Лин Б., Кальчман М.А. (август 1993 г.). «Взаимосвязь между длиной тринуклеотидного (CAG) повтора и клиническими особенностями болезни Хантингтона». Природа Генетика. 4 (4): 398–403. Дои:10.1038 / ng0893-398. PMID 8401589. S2CID 20645822.
- ^ Крафорд Д., Сноуден Дж. (2002). «Нейрописхологические и нейропсихиатрические аспекты болезни Хантингтона». В Bates G, Harper P, Jones L (ред.). Болезнь Хантингтона - Третье издание. Оксфорд: Издательство Оксфордского университета. С. 62–87. ISBN 978-0-19-851060-4.
- ^ Ди Майо Л., Сквитьери Ф, Наполитано Дж., Кампанелла Дж., Трофаттер Дж. А., Коннелли П.М. (апрель 1993 г.). «Риск суицида при болезни Гентингтона». Журнал медицинской генетики. 30 (4): 293–5. Дои:10.1136 / jmg.30.4.293. ЧВК 1016335. PMID 8487273.
- ^ а б c d е ж Харпер П. (2002). «Эпидемиология болезни Хантингтона». В Bates G, Harper P, Jones L (ред.). Болезнь Хантингтона - Третье издание. Оксфорд: Издательство Оксфордского университета. С. 159–189. ISBN 978-0-19-851060-4.
- ^ Шарон И., Шарон Р., Уилкенс Дж. П., Эрсан Т. (2010). «Деменция при болезни Хантингтона». emedicine, WebMD. Medscape. В архиве из оригинала 5 марта 2010 г.. Получено 16 мая 2010.
- ^ Драйвер-Данкли Э., Кавинесс Дж. Н. (2007). "Болезнь Хантингтона". В Schapira AH (ред.). Неврология и клиническая неврология. Мосби Эльзевьер. С. 879–885. ISBN 978-0-323-03354-1.
- ^ Эванс С.Дж., Дуглас И., Роулинз М.Д., Векслер Н.С., Тебризи С.Дж., Смит Л. (октябрь 2013 г.). «Распространенность болезни Хантингтона среди взрослых в Великобритании на основании диагнозов, зарегистрированных в общих медицинских картах». Журнал неврологии, нейрохирургии и психиатрии. 84 (10): 1156–60. Дои:10.1136 / jnnp-2012-304636. ЧВК 3786631. PMID 23482661.
- ^ Авила-Жироо Р. (1973). «Медицинские и социальные аспекты хореи Хантингтона в штате Сулия, Венесуэла». Достижения в неврологии. 1: 261–6. ISSN 0091-3952. NAID 10021247802.
- ^ а б Gusella JF, Wexler NS, Conneally PM, Naylor SL, Anderson MA, Tanzi RE, Watkins PC, Ottina K, Wallace MR, Sakaguchi AY (1983). «Полиморфный ДНК-маркер, генетически связанный с болезнью Хантингтона». Природа. 306 (5940): 234–8. Bibcode:1983Натура.306..234G. Дои:10.1038 / 306234a0. PMID 6316146. S2CID 4320711.
- ^ Сквитери Ф., Эндрю С.Е., Голдберг Ю.П., Кремер Б., Спенс Н., Цейслер Дж., Никол К., Тейлманн Дж., Гринберг Дж., Гото Дж. (Декабрь 1994 г.). «Анализ гаплотипов ДНК болезни Хантингтона раскрывает ключи к источникам и механизмам распространения CAG и причинам географических вариаций распространенности». Молекулярная генетика человека. 3 (12): 2103–14. Дои:10.1093 / hmg / 3.12.2103. PMID 7881406.
- ^ Свейнссон О., Халлдорссон С., Олафссон Э. (июль 2012 г.). «Необычно низкая распространенность болезни Хантингтона в Исландии». Европейская неврология. 68 (1): 48–51. Дои:10.1159/000337680. PMID 22722209. S2CID 207551998.
- ^ Сипиля Ю.О., Хиетала М., Сийтонен А., Пяйваринта М., Маямаа К. (январь 2015 г.). «Эпидемиология болезни Хантингтона в Финляндии». Паркинсонизм и связанные с ним расстройства. 21 (1): 46–9. Дои:10.1016 / j.parkreldis.2014.10.025. PMID 25466405.
- ^ Альмквист Э.В., Элтерман Д.С., Маклауд П.М., Хайден М.Р. (сентябрь 2001 г.). «Высокий уровень заболеваемости и отсутствие семейных историй болезни у одной четверти пациентов, у которых впервые диагностирована болезнь Хантингтона в Британской Колумбии». Клиническая генетика. 60 (3): 198–205. Дои:10.1034 / j.1399-0004.2001.600305.x. PMID 11595021. S2CID 19786394.
- ^ а б Хантингтон G (1872 г.). На хореи. Медицинский и хирургический репортер Филадельфии. 26. Гаага: Nijhoff. стр.317 –321. ISBN 978-90-6186-011-2. Получено 1 апреля 2009.
- ^ Белленир К, изд. (2004). «Болезнь Хантингтона». Справочник по генетическим заболеваниям (3-е изд.). Детройт: омниграфика. стр.159 –179. ISBN 978-0-7808-0742-6.
- ^ а б c d е ж грамм час я j Харпер П. (2002). «Болезнь Хантингтона: историческая справка». В Bates G, Harper P, Jones L (ред.). Болезнь Хантингтона - Третье издание. Оксфорд: Издательство Оксфордского университета. С. 3–24. ISBN 978-0-19-851060-4.
- ^ а б c d Векслер А., Векслер Н. (2008). Женщина, которая вошла в море. Хантингтон и создание генетической болезни. Издательство Йельского университета. п. 288. ISBN 978-0-300-10502-5. Получено 15 ноября 2015.
- ^ Лунд JC (1860). "Chorea Sti Viti i Sætersdalen. Uddrag af Distriktslæge J. C. Lunds Medicinalberetning на 1860 год". Беретнинг Ом Сундхедстилстанден: 137–138.
- ^ Lanska DJ (апрель 2000 г.). «Джордж Хантингтон (1850–1916) и наследственная хорея». Журнал истории неврологии. 9 (1): 76–89. Дои:10.1076 / 0964-704X (200004) 9: 1; 1-2; FT076. PMID 11232352. S2CID 22659368.
- ^ Броуди И.А., Уилкинс Р.Х. (сентябрь 1967 г.). «Хорея Хантингтона». Архив неврологии. 17 (3): 331. Дои:10.1001 / archneur.1967.00470270109013. PMID 4228262.
- ^ Джеллифф С.Е., Манси Э.Б., Давенпорт CB (1913). "Хорея Хантингтона: исследование наследственности". Журнал нервных и психических заболеваний. 40 (12): 796–799. Дои:10.1097/00005053-191312000-00010.
- ^ а б Давенпорт CB, Манси EB (1916). «Хорея Хантингтона применительно к наследственности и евгенике». Американский журнал безумия. 73 (2): 195–222. Дои:10.1176 / ajp.73.2.195.
- ^ Весси PR (1932). «О передаче хореи Хантингтона за 300 лет - семейная группа Бурес». Нервные и психические заболевания. 76 (6): 553–573. Дои:10.1097/00005053-193212000-00001. S2CID 147656032. Получено 1 апреля 2009.
- ^ а б Векслер AR (2002). «Хорея и община в городе девятнадцатого века». Вестник истории медицины. 76 (3): 495–527. Дои:10.1353 / bhm.2002.0150. PMID 12486915. S2CID 30791504.
- ^ Коннелли PM (май 1984 г.). «Болезнь Хантингтона: генетика и эпидемиология». Американский журнал генетики человека. 36 (3): 506–26. ЧВК 1684448. PMID 6233902.
- ^ Векслер Н.С. (2012). «Болезнь Хантингтона: пропаганда движет наукой». Ежегодный обзор медицины. 63: 1–22. Дои:10.1146 / annurev-med-050710-134457. PMID 22248319.
- ^ а б «Венесуэльский проект по болезни Хантингтона». Веб-сайт Фонда наследственных болезней. Фонд наследственных болезней. 2008. Архивировано с оригинал 10 августа 2015 г.. Получено 8 сентября 2008.
- ^ а б Макдональд М (март 1993 г.). «Новый ген, содержащий тринуклеотидный повтор, который расширяется и нестабилен на хромосомах болезни Хантингтона. Группа совместных исследований болезни Хантингтона». Клетка. 72 (6): 971–83. Дои:10.1016 / 0092-8674 (93) 90585-Е. HDL:2027.42/30901. PMID 8458085. S2CID 802885.
- ^ Бертрам Л., Танзи Р. Э. (июнь 2005 г.). «Генетическая эпидемиология нейродегенеративных заболеваний». Журнал клинических исследований. 115 (6): 1449–57. Дои:10.1172 / JCI24761. ЧВК 1137006. PMID 15931380.
- ^ La Spada AR, Roling DB, Harding AE, Warner CL, Spiegel R, Хаусманова-Петрусевич I, Yee WC, Fischbeck KH (декабрь 1992 г.). «Мейотическая стабильность и корреляция генотип-фенотип тринуклеотидного повтора при Х-сцепленной спинальной и бульбарной мышечной атрофии». Природа Генетика. 2 (4): 301–4. Дои:10.1038 / ng1292-301. PMID 1303283. S2CID 6603129.
- ^ а б c Росс Калифорния, Тебризи SJ (январь 2011 г.). «Болезнь Хантингтона: от молекулярного патогенеза к клиническому лечению». Ланцет. Неврология. 10 (1): 83–98. Дои:10.1016 / S1474-4422 (10) 70245-3. PMID 21163446. S2CID 17488174.
- ^ "Что такое HD?". Ассоциация болезни Хантингтона. Архивировано из оригинал 13 декабря 2011 г.. Получено 18 декабря 2011.
- ^ Давенпорт CB (май 1915 г.). «Хорея Хантингтона в отношении наследственности и евгеники». Труды Национальной академии наук Соединенных Штатов Америки. 1 (5): 283–5. Bibcode:1915ПНАС .... 1..283Д. Дои:10.1073 / pnas.1.5.283. ЧВК 1090803. PMID 16575999.
- ^ Роллин Б.Е. (2006). «Регулирование исследований на животных и появление этики животных: концептуальная история» (PDF). Теоретическая медицина и биоэтика. 27 (4): 285–304. Дои:10.1007 / s11017-006-9007-8. PMID 16937023. S2CID 18620094.
- ^ Doerflinger RM (февраль 2008 г.). «Проблема обмана при исследовании эмбриональных стволовых клеток». Распространение клеток. 41 (Приложение 1): 65–70. Дои:10.1111 / j.1365-2184.2008.00492.x. ЧВК 6496399. PMID 18181947.
- ^ Чепмен М.А. (июль 1990 г.). «Прогностическое тестирование на генетическое заболевание с началом у взрослых: этические и юридические последствия использования анализа сцепления для болезни Хантингтона». Американский журнал генетики человека. 47 (1): 1–3. ЧВК 1683745. PMID 2140926.
- ^ Хаггинс М., Блох М., Канани С., Куоррелл О.В., Тейлман Дж., Хедрик А., Диккенс Б., Линч А., Хайден М. (июль 1990 г.). «Этические и правовые дилеммы, возникающие при прогностическом тестировании на болезнь, начинающуюся у взрослых: опыт болезни Хантингтона». Американский журнал генетики человека. 47 (1): 4–12. ЧВК 1683755. PMID 1971997.
- ^ «Мораторий на страхование генетики продлен до 2017 года» (Пресс-релиз). Ассоциация британских страховщиков. 5 апреля 2011 г. В архиве из оригинала 4 марта 2016 г.. Получено 13 января 2016.
- ^ «Эксперт поддерживает раскрытие генного теста». Статья BBC. 7 июня 2007 г. В архиве из оригинала 26 февраля 2008 г.
- ^ Бинеделл Дж., Солдан Дж. Р., Скурфилд Дж., Харпер П.С. (ноябрь 1996 г.). «Прогностическое тестирование болезни Хантингтона: аргументы в пользу оценочного подхода к запросам подростков». Журнал медицинской генетики. 33 (11): 912–8. Дои:10.1136 / jmg.33.11.912. ЧВК 1050784. PMID 8950670.
- ^ Борри П., Гоффин Т., Нис Х, Дирикс К. (2008). «Прогностическое генетическое тестирование несовершеннолетних на генетические заболевания, развивающиеся у взрослых». Медицинский журнал Mount Sinai, Нью-Йорк. 75 (3): 287–96. Дои:10.1002 / msj.20038. PMID 18704981.
- ^ а б c Брауде PR, De Wert GM, Evers-Kiebooms G, Pettigrew RA, Geraedts JP (декабрь 1998 г.). «Неразглашение доимплантационной генетической диагностики болезни Хантингтона: практические и этические дилеммы». Пренатальная диагностика. 18 (13): 1422–6. Дои:10.1002 / (SICI) 1097-0223 (199812) 18:13 <1422 :: AID-PD499> 3.0.CO; 2-R. PMID 9949442.
- ^ "Международная Хантингтонская ассоциация". Международная Хантингтонская ассоциация. 2013. В архиве из оригинала 18 апреля 2009 г.. Получено 3 апреля 2009.
- ^ "Резолюция 531 Сената США". S. Res. 531. Сенат США. 6 апреля 2008 г. В архиве из оригинала 17 ноября 2015 г.. Получено 10 августа 2008.
- ^ а б Odling-Smee L (17 мая 2007 г.). «Биомедицинская филантропия: денежное дерево». Природа. 447 (7142): 251. Bibcode:2007Натура 447..251.. Дои:10.1038 / 447251a. S2CID 4357517.
- ^ а б «Фонд ЧДИ». chdifoundation.org. Получено 13 ноября 2020.
- ^ Отметьте E (май 2007 г.). «Биомедицинская филантропия: любовь или деньги». Природа. 447 (7142): 252–3. Bibcode:2007Натура.447..252C. Дои:10.1038 / 447252a. PMID 17507955.
- ^ а б «Болезнь Хантингтона: надежда через исследования». www.ninds.nih.gov. Получено 16 ноября 2020.
- ^ Тернер С., Шапира А.Х. (июнь 2010 г.). «Митохондриальные вещества мозга: роль в болезни Хантингтона». Журнал биоэнергетики и биомембран. 42 (3): 193–8. Дои:10.1007 / s10863-010-9290-у. PMID 20480217. S2CID 207185615.
- ^ Дикий Э.Дж., Тебризи С.Дж. (сентябрь 2014 г.). «Цели будущих клинических испытаний болезни Хантингтона: что готовится?». Двигательные расстройства. 29 (11): 1434–45. Дои:10.1002 / mds.26007. ЧВК 4265300. PMID 25155142.
- ^ а б Соломон, Шошанна (3 мая 2017 г.). «Таубе выделит 3 миллиона долларов на исследования болезни Хантингтона в США». The Times of Israel. В архиве из оригинала 3 мая 2017 г.. Получено 5 мая 2017.
- ^ а б Муньос-Санджуан I, Бейтс ГП (февраль 2011 г.). «Важность интеграции фундаментальных и клинических исследований для разработки новых методов лечения болезни Хантингтона». Журнал клинических исследований. 121 (2): 476–83. Дои:10.1172 / JCI45364. ЧВК 3026740. PMID 21285520.
- ^ Макбрайд Дж. Л., Питцер М. Р., Будро Р. Л., Дюфур Б., Хоббс Т., Охеда С. Р., Дэвидсон Б. Л. (декабрь 2011 г.). «Доклиническая безопасность РНКи-опосредованного подавления HTT у макак-резус в качестве потенциальной терапии болезни Хантингтона». Молекулярная терапия. 19 (12): 2152–62. Дои:10.1038 / мт.2011.219. ЧВК 3242667. PMID 22031240.
- ^ Кордасевич Х.Б., Станек Л.М., Ванцевич Э.В., Мазур К., Макалонис М.М., Пайтель К.А., Артатес Дж.В., Вайс А., Ченг С.Х., Шихабуддин Л.С., Хунг Г. «Устойчивое терапевтическое обращение болезни Хантингтона за счет временного подавления синтеза хантингтина». Нейрон. 74 (6): 1031–44. Дои:10.1016 / j.neuron.2012.05.009. ЧВК 3383626. PMID 22726834.
- ^ Барнс Д.В., Уитли Р.Дж. (февраль 1987 г.). «Противовирусная терапия и болезни легких». Грудь. 91 (2): 246–51. Дои:10.1172 / JCI45130. ЧВК 3026739. PMID 3026739.
- ^ «Суд над Лендмарком Хантингтоном начинается». В архиве из оригинала 21 октября 2015 г.. Получено 19 октября 2015.
- ^ «Безопасность, переносимость, фармакокинетика и фармакодинамика IONIS-HTTRx у пациентов с ранними проявлениями болезни Хантингтона - Просмотр полного текста». ClinicalTrials.gov. В архиве из оригинала 29 сентября 2015 г.. Получено 18 апреля 2016.
- ^ Wild EJ, Boggio R, Langbehn D, Robertson N, Haider S, Miller JR, Zetterberg H, Leavitt BR, Kuhn R, Tabrizi SJ, Macdonald D, Weiss A (май 2015 г.). «Количественное определение мутантного белка хантингтина в спинномозговой жидкости у пациентов с болезнью Хантингтона». Журнал клинических исследований. 125 (5): 1979–86. Дои:10.1172 / jci80743. ЧВК 4463213. PMID 25844897.
- ^ Чейз А (май 2015 г.). «Болезнь Хантингтона: спинномозговая жидкость и биомаркеры МРТ для продромального HD». Обзоры природы Неврология. 11 (5): 245. Дои:10.1038 / nrneurol.2015.63. PMID 25896083. S2CID 38300571.
- ^ Кейзер М.С., Кордасевич Х.Б., МакБрайд Д.Л. (апрель 2016 г.). «Стратегии подавления генов при доминантно наследуемых нейродегенеративных заболеваниях: уроки болезни Хантингтона и спиноцеребеллярной атаксии». Молекулярная генетика человека. 25 (R1): R53–64. Дои:10.1093 / hmg / ddv442. ЧВК 4802374. PMID 26503961.
- ^ а б Джаджадикерта, Элвин; Кешри, Свати; Павел, Марьяна; Престил, Райан; Райан, Лаура; Рубинштейн, Дэвид К. (3 апреля 2020 г.). «Индукция аутофагии как терапевтическая стратегия нейродегенеративных заболеваний». Журнал молекулярной биологии. Аутофагия при нейродегенеративных заболеваниях. 432 (8): 2799–2821. Дои:10.1016 / j.jmb.2019.12.035. ISSN 0022-2836. PMID 31887286.
- ^ Равикумар, Бринда; Вашер, Корали; Бергер, Зденек; Дэвис, Джанет Э .; Ло, Шоуцин; Oroz, Lourdes G .; Скаравилли, Франческо; Истон, Дуглас Ф .; Дуден, Райнер; O'Kane, Cahir J .; Рубинштейн, Дэвид К. (июнь 2004 г.). «Ингибирование mTOR вызывает аутофагию и снижает токсичность разложения полиглутамина в моделях болезни Хантингтона у мышей и мышей». Природа Генетика. 36 (6): 585–595. Дои:10,1038 / ng1362. ISSN 1546-1718. PMID 15146184. S2CID 7749825.
- ^ Clelland CD, Баркер Р.А., Уоттс С. (2008). «Клеточная терапия при болезни Гентингтона». Нейрохирургия. 24 (3–4): E9. Дои:10.3171 / FOC / 2008/24 / 3-4 / E8. PMID 18341412.
- ^ Cundiff PE, Anderson SA (июнь 2011 г.). «Влияние индуцированных плюрипотентных стволовых клеток на изучение заболеваний центральной нервной системы». Текущее мнение в области генетики и развития. 21 (3): 354–61. Дои:10.1016 / j.gde.2011.01.008. ЧВК 3932563. PMID 21277194.
- ^ "Поиск: болезнь Хантингтона - список результатов - ClinicalTrials.gov". Clinicaltrials.gov.
- ^ «Завершенные клинические испытания». Группа изучения Хантингтона. Архивировано из оригинал 28 июня 2012 г.. Получено 4 февраля 2012.
внешняя ссылка
- болезнь Хантингтона в Керли
- Проект НАДЕЖДА – Информационный проект Стэнфордского университета в формате HD
- HDBuzz - Новости исследований в высоком разрешении, написанные учеными простым языком
- HD Drug Works - новости о текущих курсах лечения и запланированных исследованиях
| Классификация | |
|---|---|
| Внешние ресурсы |